基于局部进化信息的DNA结合蛋白预测方法与流程
本发明属于生物信息学dna结合蛋白预测领域,具体为一种基于局部进化信息的dna结合蛋白预测方法。
背景技术:
依据序列信息鉴定dna结合蛋白是基因组注释领域最优挑战性的问题之一。dna结合蛋白在各种细胞生物学过程中起着至关重要的作用,例如基因的表达与转录。但是,使用实验方法鉴定既耗时且昂贵的。面对日益庞大的后基因组时代的海量数据,寻求一种快速且准确预测蛋白质是否为dbp的方法异常重要。
近年来,出现了许多针对dbp的预测方法,这些方法大致可以分为两类,基于结构的方法和基于序列的方法。基于结构的方法主要使用蛋白质的结构信息,例如基于蛋白质二级结构构造的α螺旋长度、氨基酸的空间组成及分子的偶极矩阵。gregoret等人开发了基于蛋白质的静电荷,电耦矩阵张量为特征的神经网络模型。张浩等人从dna结合蛋白的复杂结构中提取新的信息,在dfire能量函数的基础上引入新的体积分数校正,并深入提取蛋白质与dna之间的结合亲和力作为特征。通常,使用结构信息的同时也会使用序列信息例如idbp和dbd-hunter。例如dbd-hunter方法结合了结构对比和统计趋势的估计,在对多种蛋白质的识别预测准确率高于其他同类预测器,但此方法需要目标蛋白质的结构作为特征输入,限制了该模型的推广与应用。虽然通过结构信息进行预测方法都取得了较高的准确率,但由于蛋白质结构的数目相对于蛋白质序列的数目过少,蛋白质的结构信息获取相对滞后,这些方法都很难在后基因组时代推广。另外,由于需要预测的蛋白质往往数据库中已存在的蛋白质结构相似度较小,因此,只依赖序列信息进行预测更受青睐。
基于序列的预测方法只依赖蛋白质序列信息以预测dna结合蛋白。近年来,一系列方法被应用于预测dbp,例如:psedna-pro,idnapro-pseaac,idna-prot,local-dpp,stackdppred,k-pssm-composition,targetdbp。这些方法只需要使用序列信息以模板匹配或者机器学习的方法进行预测dbp。其通常先通过特征提取,将特征输入到支持向量机或随机森林中。例如,在idna-prot中,其使用灰色系统理论提取的伪氨基酸组合物用于代表蛋白质的成分特征,使用随机森林生成模型并进行分析预测。在local-dpp中,其使用位置特异性得分矩阵的局部进化信息作为特征,最大限度的提取局部信息,其首先将pssm进行分割为n个子矩阵,计算每段中各氨基酸进化成其他氨基酸的概率,结合子矩阵氨基酸序列,从而拼接成全局特征,用以生成预测模型。在targetdbp中,其使用氨基酸组成(aac),伪位置特异性得分矩阵等作为其特征向量,通过特征选择和差分进化组合优化不同的特征,输入至支持向量机中进行学习。
然而,多数预测模型对于局部信息提取大量参数,输入参数量庞大,从而削弱了全局信息对模型贡献度的影响。虽然在一些模型中使用算法平衡全局与局部信息的权重,但是大量无用信息使得模型规模过于庞大冗余,从而导致模型在预测效率上的不足。
技术实现要素:
本发明提出了一种基于局部进化信息的dna结合蛋白预测方法。
实现本发明目的的技术方案为:一种基于局部进化信息的dna结合蛋白预测方法,具体步骤为:
步骤1:提取蛋白质的进化信息,将进化信息分割成局部进化信息,得到用于预测的特征向量;
步骤2:使用svm-rfe+cbr特征提取方法,将步骤1中的特征向量依据其对模型的贡献度进行排序,去除无关特征;
步骤3:采用5折交叉验证方法将去除无关特征的特征向量分为5份,4份作为训练集输入svm模型对其进行训练;
步骤4:按照步骤1、2对蛋白质处理后输入步骤3得到的模型,获得预测结果。
优选地,提取蛋白质的进化信息,将进化信息分割成局部进化信息的具体方法为:
提取蛋白质的位置特异性得分矩阵;
对所述位置特异性得分矩阵进行分割,得到k个子矩阵;
对于每个子矩阵,获得其两种特征,其一:依据子矩阵序列信息统计序列中每种氨基酸进化成20种氨基酸概率之和;其二:统计子矩阵中每种氨基酸进化成20种氨基酸中的各种氨基酸概率之和;将两种特征组合得到蛋白质的序列进化特征;
将蛋白质的序列进化特征、蛋白质序列的氨基酸组成以及蛋白质二肽信息相结合,得到用于预测的特征向量。
优选地,k个子矩阵中,前k-1个子矩阵大小为
优选地,对于每个子矩阵,获得序列中每种氨基酸进化成20种氨基酸概率之和的具体方法为:
对每个子矩阵进行归一化:
其中:
式中,p’i,j为每个子矩阵中蛋白质序列s出现在第i位置的氨基酸进化成20种氨基酸中第j位置的氨基酸的概率。
统计序列中每个氨基酸进化成20种氨基酸概率之和为:
subpssmevolution=[s1s2...si...sd+u(λ)]t
其中,
优选地,所述蛋白质序列的氨基酸组成为:
其中t代表矩阵的转置,l为蛋白质序列的长度,ni为第i种氨基酸在长度为l的氨基酸序列中出现的频率。
优选地,对svm模型预测时,将预测的概率值转化为真假值与实际蛋白质是否为dna结合蛋白的真假值进行计算,得到模型评价指标;
使用网格搜索算法,通过预设svm初始参数区间范围以及模型评价指标的阈值范围,选择获得最优评价指标时的参数作为最终模型参数。
优选地,所述模型评价指标包括准确率、敏感性、特异性以及马修相关系数。
本发明与现有技术相比,其显著优点为:。
(1)模型精度的提升:本发明将多种蛋白质序列的特征相组合,结合蛋白质的局部进化信息与原进化信息和氨基酸组成、二肽信息,充分包含了蛋白质的局部与整体信息,提高了dna结合蛋白预测的计算模型的精度;
(2)模型效率的提升:本发明使用对预测精度贡献度最大的特征相组合,同时通过特征选择去除了冗余特征,使得模型的训练效率和预测效率大幅提升。
下面结合附图对本发明做进一步详细的描述。
附图说明
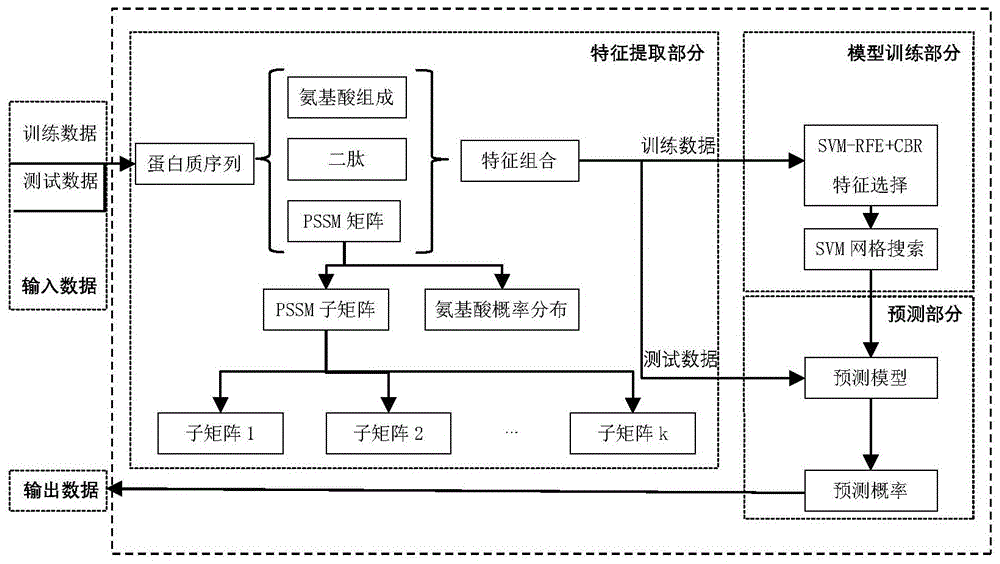
图1为本发明的流程图。
具体实施方式
一种基于局部进化信息的dna结合蛋白预测方法,包括以下步骤:
步骤1:提取蛋白质的进化信息,将进化信息分割成局部进化信息,得到用于预测的特征向量;
在某些实施例中,对于一个由n个氨基酸组成的蛋白质序列,通过psi-blast程序提取到该蛋白质的位置特异性得分矩阵pssm,其大小为n×20;再对所述位置特异性得分矩阵pssm分割,得到k个子矩阵,其中前k-1个子矩阵大小为
将蛋白质的序列进化特征与蛋白质序列的氨基酸组成和蛋白质的二肽信息三种一维向量相结合,得到用于预测的特征向量,共2620维输入特征。
步骤2:特征选择
使用svm-rfe+cbr特征提取方法,将步骤1中的特征向量依据其对模型的贡献度进行排序,去除无关特征;
步骤3:采用5折交叉验证方法将去除无关特征的特征向量分为5份,4份作为训练集输入svm模型对其进行训练;
对svm模型预测时,将预测的概率值转化为真假值与实际蛋白质是否为dna结合蛋白的真假值进行计算,得到模型评价指标;
使用网格搜索算法,通过预设svm初始参数区间范围以及模型评价指标的阈值范围,选择获得最优评价指标时的参数作为最终模型参数。
步骤4:按照步骤1、2对蛋白质处理后输入步骤3得到的模型,获得预测结果。
本发明优化了输入参数的结构,使得无效参数大幅减少,从而有效提高了训练的速度;通过svm-rfe+cbr特征筛选方法,有效提高了关键特征对模型的影响,精简特征输入同时提高预测精度。
实施例
如图1所示,本实施例中,一种基于局部进化信息的dna结合蛋白预测方法,包括以下步骤:
步骤1:特征提取
给定蛋白质序列s,其表示为s1s2s3…sl,其中si(1≤i≤l)为出现在第i位的氨基酸(残基),l是蛋白质序列s的长度。使用psi-blast获取蛋白质的进化信息pssm。pssm矩阵则是l×20(l行20列)的矩阵,其格式如下:
其中l是原蛋白质序列的长度,pi,j(i=1,2,3…l,j=1,2,3…20)是蛋白质序列中第i位进化成第j位置的氨基酸的概率得分。
通过将pssm通过行分割成k个相等的pssm矩阵,得到子矩阵公式表示为:
其中,d=(λ-1)×u(λ),表示每个子矩阵的起始序列位置在原序列的序号,u(λ)通过以下方程得出:
对每个子矩阵进行归一化:
其中:
式中,p’i,j为每个子矩阵中蛋白质序列s出现在第i位置的氨基酸进化成20种氨基酸中第j位置的氨基酸的概率。
依据归一化的子矩阵计算子矩阵的第一种特征,即统计序列中每个氨基酸进化成20种氨基酸概率之和:
subpssmevolution=[s1s2...si...sd+u(λ)]t#(6)
其中
因为第一种特征长度不定,在与子矩阵的第二种特征进行组合前,将所有子矩阵的subpssmevolution特征依次拼接则得到总长度为序列长度l的特征,由于实验数据中所有蛋白质序列的长度l均小于1000,即所有子矩阵的序列长度之和小于1000,将拼接后的subpssmevolution特征用0扩充至定长1000。
再次计算子矩阵的第二种特征。具体为将子矩阵中相同的氨基酸进化成20中不同的氨基酸的概率相加。如下所示为某一子矩阵:
在该表中,左侧第一列表示子矩阵中氨基酸序列kkespksi,第一行表示进化后的20种氨基酸,表中第二行第二列0.12表示序列中氨基酸k进化成氨基酸a的概率。以氨基酸s为例,在子矩阵的氨基酸序列中,第4位置和第7位置为氨基酸s,由表格可知,第4位置进化成氨基酸a概率为0.9,第7位置进化成氨基酸a概率为0.98,则此子矩阵中,氨基酸s进化成氨基酸a概率特征值为0.9+0.98=1.88。相似的,计算氨基酸s进化成所有20种氨基酸的概率,可获得20个特征。氨基酸s是氨基酸其中一种,分别统计20种氨基酸的特征值,可得20×20维特征。此为单个子矩阵中每种氨基酸进化成20种氨基酸中的各种氨基酸概率之和特征。则所有子矩阵共可获得20×20k维特征向量。
将蛋白质的序列进化特征中的第一种特征与第二种特征相拼接共可获得1000+20×20k维特征。本实例中取k=3,即子矩阵个数为3,则蛋白质的序列进化特征一共含2200维特征向量。
令a1a2…a19a20表示20种自然氨基酸,即氨基酸a,c,…w,y。l为蛋白质序列的长度,ni为第i种氨基酸在长度为l的氨基酸序列中出现的频率,则蛋白质的氨基酸组成特征可以用如下公式表示:
其中t代表矩阵的转置,则氨基酸组成的特征维度是20.
令fi,j代表aiaj二肽在长度为l的蛋白质中出现的频率,则蛋白质二肽特征可用如下公式表示为:
fdip=(f1,1,f1,2,…f1,20,f2,1,…f20,20)t#(8)
使用如下公式对二肽频率归一化:
其中fmax,fmin代表所有二肽频率中的最大值和最小值,二肽特征可以获得400维的特征。
由于特征均为一维向量,可以直接将蛋白质的序列进化特征与蛋白质序列的氨基酸组成和二肽信息相结合,一共可得到2200+20+400=2620维特征,以此作为输入的特征向量。
步骤2:特征选择
将所有的结果特征向量与实际真假值输入到svm-rfe+cbr中,此方法会依据各个输入向量对实际真假值的贡献度将输入特征进行打分并给出预测结果真或假。首先去除打分值为0的无关输入向量,留下323个位置的有效输入变量。由于svm-rfe+cbr无法自动确定最佳使用特征数量,使用选择前n(10<n<324)个得分最高的有效输入变量分别进行实验,依据每个n值得预测结果与实际结果计算对应于每个n的auc值,当n取157时得到最优auc值。即确定157个输入向量用于模型训练。
步骤3:模型训练
使用5折交叉验证,通过将训练数据分为5份,其中一份作为训练用测试集,从而在训练中充分利用训练集中的各个数据。选取训练集中进行特征选择后的157维向量作为svm模型训练(使用径向基函数内核)的输入数据,可以得到对于每个输入数据的预测为真的概率值;
使用阈值分割的方法,将预测的概率值转化为真假值与实际蛋白质是否为dna结合蛋白的真假值进行计算从而得到acc(准确率),se(敏感性),sp(特异性),mcc(马修相关系数)模型评价指标。
在最优化svm模型训练算法的参数时,使用网格搜索算法,通过预设svm初始参数区间范围以及阈值分割方法中的模型评价指标阈值范围,选择获得最优准确率时的参数作为最终模型参数。
在实际进行预测时,下表为本实施例与其他现有方法在基准数据集pdb1075进行训练预测对比结果:
从表中可以看出相对于既有方法,本发明在训练用时缩短的同时,模型评价指标acc,se,mcc均有所提升。
步骤4:按照步骤1、2对蛋白质处理后输入步骤3得到的模型,获得预测结果。
- 还没有人留言评论。精彩留言会获得点赞!