检测基因组结构变异的方法、计算设备和存储介质与流程
1.本发明概括而言涉及生物信息领域,并且具体地,涉及一种用于检测基因组结构变异的方法、计算设备和计算机存储介质。
背景技术:
2.结构变异(structure variation, sv)是指基因组中长度在50bp(碱基)及以上的缺失(deletion)、插入(insertion)、重复(duplication)、倒位(inversion)和易位(translocation)。结构变异是人类基因组重要的变异来源,与种群多样性、进化、遗传疾病和肿瘤有密切的关系。
3.与单核苷酸变异(snv)和小的indel(小于50bp的插入和缺失)相比,结构变异涉及的二代序列比对情况更复杂,比对准确性更低。目前,已经有多种序列比对信息用于结构变异的检测,包括序列比对内部的插入和缺失、切分比对(split
‑
mapping)、软剪切(soft
‑
clipped)、异常配对序列(discording paired reads)、序列边缘比对质量低、双端序列单端比对(配对的两条序列中仅1条可以很好地比对到参考基因组)。切分比对是指一条测序序列比对到参考基因组时被切分为多段比对到参考基因组上的情况。软剪切是指测序序列比对到参考序列时,末端部分子序列比对到参考基因组的其他位置或没有比对到参考基因组。bam文件使用cigar记录软剪切,例如cigar:10s140m 表示序列的头部有10个碱基发生了软剪切。
4.根据是否使用组装来检测结构变异,将结构变异检测软件分为两类。第一类结构变异检测软件不具有组装特性,该类可以利用上述比对信息中的几种,例如delly使用基于图论的最大团查找算法对候选变异进行聚类和对单条序列中未比对的部分序列进行重新比对来检测结构变异,lumpy提供了一种概率模型对候选变异进行聚类和推测最可能的断点位置。由于缺乏组装功能,第一类结构变异检测软件检测插入变异的能力低下,检测的结构变异断点准确性较差。第二类结构变异检测软件具有组装特性,这类结构变异检测软件尽可能多的使用上述比对信号,同时使用了局部组装的方法检测结构变异,例如manta、svaba、gridss。第二类结构变异检测软件相对第一类软件,提升了插入变异的检测能力和检测结构变异断点的准确性。
5.结构变异主要由插入和缺失组成,其他类型的结构变异比较稀有。与检测缺失相比,使用二代测序技术检测插入更加困难。首先,切分比对可以指示长片段序列缺失,但是当序列插入的长度接近测序读长时切分比对指示序列插入的能力下降为0。其次,插入片段长度(insert size)异常的配对序列可以用于检测序列缺失,但是无法找到具有统计差异的指示序列插入的插入片段长度异常的配对序列。最后,即使使用局部组装算法,当插入序列的长度接近2倍的测序文库插入片段长度时,插入的检出能力接近0。插入片段是指测序的目标dna/rna片段,测序之前通常需要对dna/rna序列进行片段化然后在两端加上接头(adaptor),因此将两个接头中间的目标dna/rna片段称为插入片段。二代双端测序(paired
‑
end sequencing)技术从插入片段的两端进行检测,得到配对(paired)的两条序
列。插入片段长度是指插入片段的长度。插入片段的实际物理长度在长度大于配对序列长度之和时无法直接通过测序数据得到。然而,将配对序列比对到参考基因组后,可以通过计算配对序列两端的距离计算插入片段长度,发生结构变异时可能会导致计算得到的插入片段长度异常。异常配对是指计算得到的插入片段长度过长或者配对序列的方向异常(不是正向
‑
反向配对)。
6.以manta为例,通过输入标准比对文件,manta可以发现、组装、检测结构变异并对结构变异打分。manta的工作流程主要分为两步:首先构建全基因组断点末端图,然后处理图中的组件(节点,边)以生成结构变异假设、组装、打分和以vcf格式报告结构变异。
技术实现要素:
7.然而,当前的上述结构变异检测方法对于插入变异的检测性能差、敏感度低,特别是对长片段插入变异的检测敏感度低,并且结构变异的整体检测性能低。
8.针对上述问题中的至少一个,本发明提供了一种检测基因组结构变异的方案,其能够对基因组序列(一条或者多条染色体,或者全基因组)中的结构变异信号发现、组装、打分并输出检测结果(如以标准的vcf格式)。
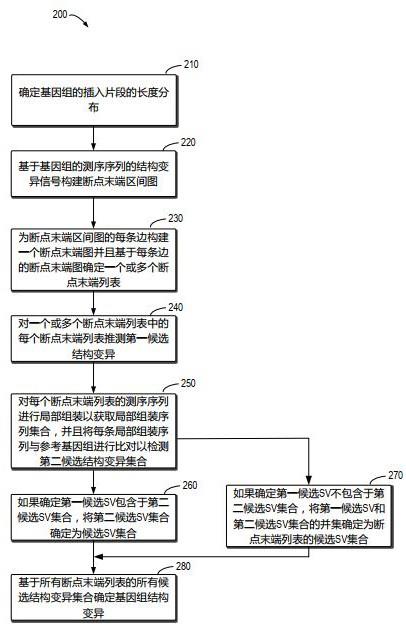
9.根据本发明的一个方面,提供了一种检测基因组结构变异的方法。该方法包括:基于所述基因组的测序序列的结构变异信号构建断点末端区间图;为所述断点末端区间图的每条边构建一个断点末端图并且基于每条边的断点末端图确定一个或多个断点末端列表,每个断点末端列表聚类了具有相同结构变异类型的断点末端;对所述一个或多个断点末端列表中的每个断点末端列表推测第一候选结构变异;对所述一个或多个断点末端列表中的每个断点末端列表的测序序列进行局部组装以获取所述断点末端列表的局部组装序列集合,并且将每条局部组装序列与参考基因组进行比对以检测第二候选结构变异集合;响应于确定所述第一候选结构变异包含于所述第二候选结构变异集合中,将所述第二候选结构变异集合确定为所述断点末端列表的候选结构变异集合;响应于确定所述第一候选结构变异不包含于所述第二候选结构变异集合中,将所述第一候选结构变异和所述第二候选结构变异集合的并集确定为所述断点末端列表的候选结构变异集合;以及基于所有断点末端列表的所有候选结构变异集合确定所述基因组结构变异。
10.根据本发明的另一个方面,提供了一种计算设备。该计算设备包括:至少一个处理器;以及至少一个存储器,该至少一个存储器被耦合到该至少一个处理器并且存储用于由该至少一个处理器执行的指令,该指令当由该至少一个处理器执行时,使得该计算设备执行根据上述方法的步骤。
11.根据本发明的再一个方面,提供了一种计算机可读存储介质,其上存储有计算机程序代码,该计算机程序代码在被运行时执行如上所述的方法。
12.在一种实施例中,其中基于所述基因组的测序序列的结构变异信号构建断点末端区间图包括:对所述基因组中的每条染色体并行地在标准比对文件中查找所述测序序列的结构变异信号,并保存所述结构变异信号的测序序列;将所述结构变异信号转换为断点末端以构建所述的断点末端区间图,其中所述断点末端区间图包括一个或多个第一节点和一个或多个第一边,每个第一节点为一个断点末端区间,并且每个第一边连接两个第一节点或者同一第一节点;以及基于所述断点末端区间图的每个第一边的权值和结构变异信号的
数目对所述断点末端区间图的一个或多个第一边进行过滤,其中每个第一边的权值由与所述第一边对应的断点末端的类型、每种类型的断点末端的数目和所述断点末端支持的结构变异长度确定。
13.在一种实施例中,其中对所述基因组中的每条染色体并行地在标准比对文件中查找所述测序序列的结构变异信号还包括:在所述基因组包括至少两条染色体的情况下,确定与一条染色体上的比对记录相对应的配偶比对记录是否位于另一条染色体上;以及响应于确定与一条染色体上的比对记录相对应的配偶比对记录位于另一条染色体上,生成染色体间易位类型的结构变异信号。
14.在一种实施例中,其中为所述断点末端区间图的每条边构建一个断点末端图并且基于每条边的断点末端图确定一个或多个断点末端列表包括:对于所述断点末端区间图的每条第一边,构建一个断点末端图,所述断点末端图包括一个或多个第二节点和一个或多个第二边,每个第二节点为一个断点末端,并且每个第二边连接断点末端具有至少部分重叠的两个第二节点;基于每个断点末端图中的第二节点之间的关联性,将每个断点末端图分为一个或多个独立子图;以及对每个独立子图进行聚类以产生一个或多个类,每个类包含一个断点末端列表。
15.在一种实施例中,其中对所述一个或多个断点末端列表中的每个断点末端列表推测第一候选结构变异包括:确定所述断点末端列表支持的结构变异类型作为所述第一候选结构变异的结构变异类型;确定所述断点末端列表支持的结构变异长度的中值作为所述第一候选结构变异的长度;以及确定所述断点末端列表支持的结构变异位置的中值作为所述第一候选结构变异的位置。
16.在一种实施例中,其中对所述一个或多个断点末端列表中的每个断点末端列表的测序序列进行局部组装以获取所述断点末端列表的局部组装序列集合包括:确定所述断点末端列表中的所有断点末端各自的测序序列;基于所述断点末端列表中的所有断点末端的所有测序序列和前一循环获取的所述断点末端列表的局部组装序列集合构建输入序列集合,并且以大小为第一值、步长为一的滑动窗依次获取所述输入序列集合中的碱基片段;基于获取的碱基片段构建德布莱英图,其中所述德布莱英图包括多个第三节点和多个第三边,每个第三节点为一个碱基片段,每个第三边连接相邻的两个碱基片段;以及基于所述德布莱英图中的多个第三节点的碱基片段之间的重叠关系获取所述局部组装序列集合。
17.在一种实施例中,该方法还包括:将所述第一值增大预定值;确定增大后的第一值是否小于或等于第二值;响应于确定增大后的第一值小于或等于第二值,基于所述断点末端列表中的所有断点末端的所有测序序列和所述局部组装序列集合构建用于下一循环的输入序列集合,并且在下一循环中以大小为增大后的第一值、步长为一的滑动窗查找所述输入序列集合中的碱基片段。
18.在一种实施例中,其中基于获取的碱基片段构建德布莱英图包括:基于获取的碱基片段构建初始德布莱英图;对所述初始德布莱英图执行以下操作中的至少一种以构建所述德布莱英图:1)检测所述初始德布莱英图以确定是否存在环,如果存在环则跳过本次循环;2)检测所述初始德布莱英图以确定是否存在泡状结构,其中所述泡状结构具有两条路径,所述两条路径具有相同的开始第三节点和结束第三节点,如果确定存在泡状结构,则基于所述两条路径上的第三节点的数量和第三边的权重从所述初始德布莱英图中去除所述
两条路径中权重之和较低的一条路径;3)检测所述初始德布莱英图以确定是否存在弱末梢,其中所述弱末梢表示每个第三节点数目小于第一阈值并且每个第三边的权值低于第二阈值的末端节点自由路径,如果确定存在所述弱末梢,则从所述初始德布莱英图中去除所述弱末梢;以及4)检测所述初始德布莱英图以确定是否存在分支状的头和尾,如果确定存在分支状的头,则合并所述初始德布莱英图中的分支状的头,如果确定存在分支状的尾,则合并所述初始德布莱英图中的分支状的尾,其中头表示从入度为0的节点开始的路径,尾表示以出度为0的节点结束的路径,分支状的头表示连接到同一节点的数目至少为2的头的集合,分支状的尾表示连接到同一节点的数目至少为2的尾的集合。
19.在一种实施例中,其中所述碱基片段包括独有碱基片段和非独有碱基片段,其中独有碱基片段指示对于所述输入序列集合中的任意一条输入序列来说,在该输入序列中未重复出现的碱基片段,非独有碱基片段指示在该输入序列中重复出现的碱基片段。其中基于获取的碱基片段构建德布莱英图还包括:确定一个碱基片段是否存在于所述德布莱英图中;响应于确定一个碱基片段不存在于所述德布莱英图中,将所述碱基片段作为一个第三节点添加到所述德布莱英图中,并且在所述碱基片段和所述碱基片段的前一碱基片段之间添加第三边;响应于确定一个碱基片段存在于所述德布莱英图中,确定所述碱基片段是独有碱基片段还是非独有碱基片段;响应于确定所述碱基片段是独有碱基片段,更改所述独有碱基片段与所述独有碱基片段的前一碱基片段之间的第三边的权重;响应于确定所述碱基片段是非独有碱基片段,获取所述非独有碱基片段与所述非独有碱基片段的前一碱基片段之间的第三边;确定是否获取到所述非独有碱基片段与所述非独有碱基片段的前一碱基片段之间的第三边;响应于获取到所述非独有碱基片段与所述非独有碱基片段的前一碱基片段之间的第三边,更改所述非独有碱基片段与所述非独有碱基片段的前一碱基片段之间的第三边的权重;以及响应于没有获取到所述非独有碱基片段与所述非独有碱基片段的前一碱基片段之间的第三边,将所述非独有碱基片段作为一个第三节点添加到所述德布莱英图中,并且在所述非独有碱基片段和所述非独有碱基片段的前一碱基片段之间添加第三边。
20.在一种实施例中,其中基于所有断点末端列表的所有候选结构变异集合确定所述基因组结构变异包括:使用贝叶斯方法,基于所述候选结构变异集合中的每个候选结构变异的每种基因型的先验概率和在该基因型的条件下出现观测比对数据的条件概率确定所述候选结构变异的每种基因型的后验概率;基于所述候选结构变异的每种基因型的后验概率确定所述候选结构变异的每种基因型的似然性分值;基于所述候选结构变异的每种基因型的似然性分值,将似然性分值最小的基因型确定为所述候选结构变异的基因型;基于所述候选结构变异的每种基因型的似然性分值,将纯合参考基因型的似然性分值确定作为所述候选结构变异的质量值;确定所述候选结构变异的质量值是否大于预定值;响应于确定所述候选结构变异的质量值大于所述预定值,将所述候选结构变异作为所述断点末端列表的结构变异;以及合并所有断点末端列表的结构变异以获取所述基因组结构变异。
21.在一种实施例中,该方法还包括确定所述基因组的插入片段的长度分布。其中确定所述基因组的插入片段的长度分布包括:获取所述基因组的标准比对文件,所述标准比对文件包括所述基因组的测序序列与参考基因组之间的比对记录;对所述参考基因组的每条染色体,不重复地随机抽取第一数量个染色体区间,其中每个染色体区间大小相同并且
和“一些实施例”表示“至少一个示例实施例”。术语“另一实施例”表示“至少一个另外的实施例”。术语“第一”、“第二”等等可以指代不同的或相同的对象。
37.如前所述,当前的结构变异检测方法对于插入变异的检测性能差、敏感度低,特别是对长片段插入变异的检测敏感度低,并且结构变异的整体检测性能低。
38.针对上述问题中的至少一个,本发明提供了一种用于检测基因组结构变异的方案。首先,基于待检测的基因组的测序序列的结构变异信号构建断点末端区间图。为该断点末端区间图的每条边构建一个断点末端图并且基于每条边的断点末端图确定一个或多个断点末端列表,每个断点末端列表聚类了具有相同结构变异类型的断点末端。然后,对每个断点末端列表推测第一候选结构变异。对一个或多个断点末端列表中的每个断点末端列表进行局部组装以获取该断点末端列表的局部组装序列集合,并且将每条局部组装序列与参考基因组进行比对以检测第二候选结构变异集合。如果确定第一候选结构变异包含于第二候选结构变异集合,将第二候选结构变异集合确定为所述断点末端列表的候选结构变异集合;如果确定第一候选结构变异不包含于第二候选结构变异集合,将第一候选结构变异和第二候选结构变异集合的并集确定为所述断点末端列表的候选结构变异集合。最后,基于所有断点末端列表的所有候选结构变异集合确定该基因组结构变异。
39.利用上述方案,能够提升基因组结构变异的整体检测性能、插入变异的检测性能以及插入变异的检测敏感性。
40.图1示出了根据本发明的实施例的用于检测基因组结构变异的设备100的示意图。如图1中所示,设备100可以包括断点末端区间图构建单元121、断点末端列表确定单元122、第一候选结构变异确定单元123、第二候选结构变异集合确定单元124、候选结构变异集合确定单元125和结构变异确定单元126。断点末端区间图构建单元121用于基于待检测的基因组的测序序列的结构变异信号构建断点末端区间图。断点末端列表确定单元122用于为断点末端区间图的每条边构建一个断点末端图并且基于每条边的断点末端图确定一个或多个断点末端列表。第一候选结构变异确定单元123用于基于基因组的插入片段的长度分布推测每个断点末端列表的第一候选结构变异。第二候选结构变异集合确定单元124用于对一个或多个断点末端列表中的每个断点末端列表进行局部组装以获取该断点末端列表的局部组装序列集合,并且将每条局部组装序列与参考基因组进行比对以检测第二候选结构变异集合。候选结构变异集合确定单元125用于确定每个断点末端列表的候选结构变异集合。结构变异确定单元126用于基于所有断点末端列表的所有候选结构变异集合确定待检测的基因组的结构变异。断点末端区间图构建单元121、断点末端列表确定单元122、第一候选结构变异确定单元123、第二候选结构变异集合确定单元124、候选结构变异集合确定单元125和结构变异确定单元126的具体功能将在下面参考图2至图8进行描述。
41.在一些实施例中,设备100还可以包括插入片段长度分布确定单元110,用于确定基因组的插入片段的长度分布。虽然图1中将插入片段长度分布确定单元110显示为设备100的一部分,但是本领域技术人员可以理解,插入片段长度分布确定单元110可以位于设备100之外,即设备100可以获取由其他外部设备确定的插入片段的长度分布。
42.设备100可以包括至少一个处理器和与该至少一个处理器耦合的至少一个存储器,该存储器中存储有可由该至少一个处理器执行的指令,该指令在被该至少一个处理器执行时执行如下所述的方法200的至少一部分。断点末端区间图构建单元121、断点末端列
表确定单元122、第一候选结构变异确定单元123、第二候选结构变异集合确定单元124、候选结构变异集合确定单元125、结构变异确定单元126和插入片段长度分布确定单元110中的至少一部分可以实现为单独的硬件(如芯片),或者可以分别由上述指令的一部分来实现为软件形式。设备100的具体结构例如可以如下结合图9所述。
43.图2示出了根据本发明的实施例的用于检测基因组结构变异的方法200的流程图。方法200可以由图1中所示的设备100执行。
44.如图2中所示,在步骤220,设备100(如断点末端区间图构建单元121)可以基于待检测的基因组的测序序列的结构变异信号构建该基因组的断点末端区间图。
45.在本文中,待检测的基因组的测序序列可以是检测对象的全基因组序列(所有染色体),或者是全基因组序列的一部分,也可以是检测对象的一条或多条染色体。
46.测序序列的结构变异信号可以是从标准比对文件中查找得到的,其指示待检测的测序组的测序序列与参考基因组的比对结果,可以用作后续的结构变异检测的证据。
47.标准比对文件是通过基因检测领域的各种比对软件(如bwa
‑
mem)得到的基因组的测序序列与参考基因组的比对结果信息(即比对记录)。常见的比对文件格式例如包括sam和bam。sam的全称是sequence alignment/map format(序列对齐/映射格式),其是一种序列比对格式标准,用于表示测序序列映射到基因组上的比对结果或者任意的多重比对结果。bam是sam的二进制形式。
48.断点末端(breakend,bnd)是指结构变异的断裂点(即断点)的末端位置。在用于描述结构变异数据的文件(如vcf(variant call format,变异检测格式)文件)中,断点末端可以是一个描述结构变异的字符串。在结构变异检测软件中,断点末端是一种抽象的数据结构,用于表示断点的位置等信息。断点末端区间(breakend region)是将结构变异的断点末端的位置向前后各延伸一定的距离所形成的区间(region或interval)。对于不同的结构变异信号,延伸的距离可以不同,通常可以在10
‑
300bp之间。
49.图3示出了根据本发明一种实施例的用于构建断点末端区间图的步骤220的流程图。
50.如图3中所示,步骤220可以包括子步骤222,其中对该基因组中的每条染色体并行地在标准比对文件中查找该测序序列的结构变异信号,并保存查找得到的结构变异信号的测序序列。其中,结构变异信号例如可以是在上述sam或bam文件中查找的,并且查找到的结构变异信号的测序序列可以保存到诸如sqlite3的数据库中。
51.结构变异信号的类型可以包括如下几种中的至少一部分:1. align,序列比对内部的indel;2. pair,异常配对的双端序列;3. split,切分比对;4. edge,部分软剪切的(soft
‑
clipped)或部分比对质量差;5. hang,双端序列单端比对。
52.这些结构变异信号可以用于确定如下结构变异类型中的一种或几种:缺失(del)、插入(ins)、重复(dup)、倒位(inv)、染色体间易位(tra)、未知(unknown)。
53.根据edge和hang类型的结构变异信号无法得知相应的结构变异类型,因此将edge和hang类型的结构变异信号支持的结构变异类型设为“未知”。“未知”类型的结构变异将不
会输出到最终的结果中。
54.具体方法如下:align类型的结构变异信号检测方法如下:第一,对比对记录进行过滤,去除未比对的(unmapped)比对记录,去除重复的(duplicated)比对记录,去除次要的(secondary)比对记录,去除比对分数(mapping score)小于最低比对分数阈值(如15)的比对记录;第二,根据比对记录的cigar、比对位置、测序序列获取插入和缺失信号。这里,cigar是标准比对文件中由长度和操作构成的字符串序列,用于表示两条序列比对时序列对齐(aligned)、序列插入(insertion)、序列缺失(deletion)等情况。例如:10m2i40m2d30m为一个cigar字符串,操作由字母表示,操作的长度由数字表示并置于操作之前。
55.pair类型的结构变异信号检测方法如下:第一,对比对记录进行过滤,去除配对序列都未比对的(unmapped)比对记录,去除重复的(duplicated)比对记录,去除次要的(secondary)比对记录,去除非配对比对记录(not paired),去除补充(supplementary)比对记录;第二,如果配对序列比对到同一染色体且比对记录配对方向正常(正向
‑
反向组合)且插入片段长度大于插入片段长度阈值i
max
(i
max
=i
m
+4i
δ
,其中i
m
和i
δ
分别为本次测序的插入片段的长度分布的中值和标准差),那么生成结构变异类型为“缺失”的结构变异信号;第三,如果配对序列比对到同一染色体且比对记录的配对方向为反向
‑
正向组合,那么生成结构变异类型为“重复”的结构变异信号;第四,如果配对序列比对到同一染色体且比对记录的配对方向为正向
‑
正向组合或负向
‑
负向组合,那么生成结构变异类型为“倒位”的结构变异信号;第五,如果比对记录的配偶序列比对到其他染色体,那么生成结构变异类型为“染色体间易位”的结构变异信号;最后,对检测的pair类型信号进行补充处理,如果配对序列的比对分数都小于最低比对分数阈值那么将其去除,如果配对序列中的一条记录的比对分数小于最低比对分数阈值,另一条记录的比对分数大于或等于最低比对分数阈值那么将pair类型的结构变异信号转换为hang类型的结构变异信号。
56.split类型的结构变异信号检测方法如下:第一,对比对记录进行过滤,去除未比对(unmapped)比对记录,去除重复的(duplicated)比对记录,去除次要的(secondary)比对记录,去除补充(supplementary)的比对记录,去除比对分数(mapping score)小于最低比对分数阈值(如15)的比对记录,去除不是切分比对的比对记录(不含有sa(supplementary alignments)标签)以得到主比对记录;第二,从sa标签中获取补充比对记录,对补充比对记录进行过滤,去除比对分数小于最低比对分数阈值的补充比对记录,如果补充比对记录的数目不等于1,那么中止split类型的结构变异信号查找;第三,如果主比对记录和补充比对记录在同一条染色体上且比对方向一致,计算主比对记录和切分比对记录在测序序列上的距离d
seq
和在参考基因组上的距离d
ref
,如果d
ref
‑
d
seq
≥15,那么生成结构变异类型为“缺失”的结构变异信号,如果d
seq
‑
d
ref
≥15,那么生成结构变异类型为“重复”或者“插入”的结构变异信号;第四,如果主比对记录和补充比对记录在同一条染色体上但比对方向相反,那么生成结构变异类型为“倒位”的结构变异信号;第五,如果主比对记录和补充比对记录在不同的染色体,那么生成结构变异类型为“染色体间易位”的结构变异信号。
57.edge类型的结构变异信号检测方法如下:第一,对比对记录进行过滤,去除未比对的(unmapped)比对记录,去除重复的(duplicated)比对记录,去除次要的(secondary)比对记录,去除补充(supplementary)的比对记录,去除比对分数(mapping score)小于最低比
对分数阈值(如15)的比对记录;第二,在比对记录的两端分别查找连续5个碱基及以上完全匹配的第一个位置,计算该位置与测序序列末端的距离d,如果d大于或等于8,那么生成edge类型的结构变异信号。
58.hang类型的结构变异信号检测方法如下:第一,对比对记录进行过滤,去除配对序列都未比对的(unmapped)比对记录,去除重复的(duplicated)比对记录,去除次要的(secondary)比对记录,去除补充(supplementary)的比对记录,去除非配对序列(not paired);第二,如果比对记录未比对或者比对记录的配偶序列未比对,那么生成hang类型的结构变异信号。需要说明的是,配对序列中的一条序列的比对分数低于阈值的hang类型信号由pair类型的信号转换而来。
59.其中,可以看出,在待检测的基因组包括至少两条染色体的情况下,子步骤222中还可以确定与一条染色体上的比对记录相对应的配偶比对记录是否位于另一条染色体上;以及在确定与一条染色体上的比对记录相对应的配偶比对记录位于另一条染色体上,生成染色体间易位类型的结构变异信号。
60.继续图3,接下来,在子步骤224,设备100将子步骤222获取的结构变异信号转换为断点末端以构建待检测的基因组的测序序列的断点末端区间图。
61.根据断点末端支持的结构变异类型(“插入”,“缺失”,“重复”,“倒位”,“易位”,“未知”),一个断点末端具有1个断点末端区间或2个断点末端区间。具体的,“插入”和“未知”类型的断点末端只有1个断点末端区间,“缺失”、“重复”、“倒位”和“易位”类型的断点末端有2个断点末端区间。断点末端区间图中的每个节点为一个合并的断点末端区间。对每个断点末端,根据它的断点末端区间在参考基因组上的位置排序,位置较小的断点末端区间标记为断点末端区间1,位置较大的断点末端区间标记为断点末端区间2,只有1个断点末端区间的直接标记为断点末端区间1。对所有的断点末端,按照区间重叠分别对断点末端区间1和断点末端区间2分别进行聚类和合并,得到合并的断点末端区间1的集合和合并的断点末端区间2的集合,记录与每个合并的断点末端区间关联的断点末端标签。将所有合并的断点末端区间1作为节点加入到断点末端区间图中,将所有合并的断点末端区间2作为节点加入到断点末端区间图中。对每个合并的断点末端区间1的节点查找其包含的断点末端的标签是否也存在于合并的断点末端区间2的节点中,如果存在,那么在两个节点之间新增边或增加边的权值,如果不存在那么对节点增加自连边(self
‑
edge)或增加自连边的权值。为了与下文所述的断点末端图和dbg进行区分,将断点末端区间图中的节点称为第一节点,将断点末端区间图中的边称为第一边,该断点末端区间图包括一个或多个第一节点和一个或多个第一边,每个第一节点为一个断点末端区间,并且每个第一边连接两个第一节点或者同一第一节点。
62.然后,在子步骤226,设备100可以基于该断点末端区间图的每个第一边的权值和结构变异信号的数目对该断点末端区间图的一个或多个第一边进行过滤。每个第一边的权值由与该第一边对应的断点末端的类型、每种类型的断点末端的数目和该断点末端支持的结构变异长度确定。
63.如前所述,结构变异信号的类型可以包括align、pair、split、edge、hang中的至少一个,这里使用这些结构变异信号的类型来表示对应的断点末端的类型。在一种实施例中,可以将align、pair、split、edge、hang类型的断点末端的权重分别设置为不同值。在另一种
实施例中,可以将align、pair、split、edge、hang中的一些类型的断点末端的权重设置为一个值,将其中的另一些类型的断点末端的权重设置为另一个值。例如,可以将align、split和pair类型的断点末端的权重设置为相同的值w1,将edge和hang类型的断点末端的权重设置为另一相同的值w2。权重w1可以大于权重w2,例如可以将权重w1设置为3,将权重w2设置为2。
64.如果一个边的断点末端支持的结构变异长度大于预定值(例如大于嵌合长度(1m bp)),则可以将基于该断点末端支持的结构变异长度的权重减少1。
65.每个第一边的权值通过计算与该第一边对应的每个断点末端的权值之和来确定。
66.对该断点末端区间图的第一边进行过滤可以包括从该断点末端区间图的第一边中选择权值大于或等于预定值(如8)并且结构变异信号的数目小于另一预定值(如500)的第一边。通过这样的过滤,能够显著去除低深度和高复杂度的第一边。
67.返回图2,在步骤230,设备100(例如断点末端列表确定单元122)可以为该断点末端区间图的每条边构建一个断点末端图并且基于每条边的断点末端图确定一个或多个断点末端列表,每个断点末端列表聚类了具有相同结构变异类型的断点末端。
68.与断点末端区间图类似,断点末端图是由断点末端构成的图,断点末端图中的每个节点为一个断点末端,设置一种断点末端关联的规则,在具有关联的断点末端之间新增边。为了与上文所述的断点末端区间图和下文所述的dbg进行区分,将断点末端图中的节点称为第二节点,将断点末端图中的边称为第二边,该断点末端图包括一个或多个第二节点和一个或多个第二边,每个第二节点为一个断点末端。
69.根据本发明的一种实施例,断点末端关联的规则如下:首先,断点末端支持的结构变异类型必须一致;然后,对于“缺失”,“重复”,“倒位”类型的断点末端,如果两个断点末端支持的结构变异的区间有重叠,那么这两个断点末端存在关联,否则不存在;对于“插入”和“未知”类型的断点末端,如果两个断点末端之间的距离小于预设值,那么这两个断点末端存在关联,否则不存在;对于“易位”类型的断点末端,如果两个断点末端的第一距离(位置1之间的距离)和第二距离(位置2之间的距离)都小于预设值,那么这两个断点末端存在关联,否则不存在。
70.图4示出了根据本发明一种实施例的确定断点末端列表的步骤230的流程图。
71.如图4中所示,步骤230可以包括子步骤232,其中对于断点末端区间图的每条第一边,构建一个断点末端图。
72.接下来,在子步骤234,可以基于每个断点末端图中的第二节点之间的关联性,将每个断点末端图分为一个或多个独立子图。如前所述,断点末端图中的每个第二边连接存在至少部分重叠的两个第二节点,即,支持的结构变异相似。因此,可以根据断点末端图中的第二节点之间的关联性将其分为一个或多个独立子图,每个独立子图所包含的断点末端至少属于同一种类型的结构变异。
73.在一种实施例中,可以利用并查集(union find set)算法来将断点末端图分为一个或多个独立子图。并查集是一种维护各个不相交集合之间的关系的数据结构,可以用于维护图的连通分量或是变量之间的关系。
74.接下来,在子步骤236,对子步骤234获得的每个独立子图进行聚类以产生一个或多个类,每个类包含一个断点末端列表。
75.在一种实施例中,可以利用标签传播算法(label
‑
propagation
‑
algorithm,lpa)对每个独立子图进行聚类。lpa是一种基于标签传播的局部社区划分算法。对于网络中的每一个节点,在初始阶段,标签传播算法对于每一个节点都会初始化一个唯一的标签。每一次迭代都会根据与自己相连的节点所属的标签更新自己的标签,更新的原则是选择与其相连的节点中所属标签最多的节点标签作为自己的节点标签,即标签传播。随着节点标签不断传播,最终,连接紧密的节点将有相同的标签,从而可以根据所属的标签将图中的节点进行聚类。
76.更具体地,对于一个独立子图来说,可以首先为该独立子图中的每个第二节点分配一个不同的唯一标签,通过上述标签传播算法对该独立子图中的所有第二节点进行聚类,将最终具有相同标签的第二节点归为一个类,该类包含具有相同标签的这些第二节点的断点末端。
77.注意,这里虽然以标签传播算法为例对每个独立子图的聚类进行了描述,但是本领域技术人员可以理解,本发明并不局限于标签传播算法,而是可以使用其他的聚类算法,如层次聚类,louvain模块度算法等。
78.在步骤230中所述的局部断点末端图聚类方法中,通过使用诸如标签传播算法的聚类算法进行结构变异断点末端聚类,解决了现有技术中单纯基于重叠对断点末端聚类容易受稀有错误断点末端影响的问题,聚类结果更加准确。
79.继续图2,接下来,在步骤240,设备100(例如第一候选结构变异确定单元123)对步骤230中确定的一个或多个断点末端列表中的每个断点末端列表,基于待检测的基因组的插入片段的长度分布推测第一候选结构变异。
80.具体地,设备100可以确定该断点末端列表支持的结构变异类型作为第一候选结构变异的结构变异类型;确定该断点末端列表支持的结构变异长度的中值作为第一候选结构变异的长度;以及确定该断点末端列表支持的结构变异位置的中值作为第一候选结构变异的位置。
81.此外,在推测第一候选结构变异时,还可以从该断点末端列表中去除结构变异长度异常的断点末端和结构变异位置异常的断点末端。例如,结构变异长度或结构变异位置的概率密度分布中概率值在0至q1(如0.025)和q2(如0.975)至1之间的值被视为异常值。
82.继续图2,接下来,在步骤250,设备100(例如第二候选结构变异集合确定单元124)对步骤230中确定的一个或多个断点末端列表中的每个断点末端列表的测序序列进行局部组装以获取该断点末端列表的局部组装序列集合(contigs),并且将每条局部组装序列(contig)与参考基因组进行比对以检测第二候选结构变异集合。
83.图5示出了根据本发明一种实施例的在步骤250中获取断点末端列表的局部组装序列集合的子步骤的流程图。
84.如图5中所示,步骤250可以包括子步骤251,其中确定该断点末端列表中的所有断点末端各自的测序序列。在一些实施例中,一个断点末端的测序序列可以是从上述标准比对文件中查找的。在另一些实施例中,一个断点末端的测序序列可以是从上述步骤220中所保存的诸如sqlite3的数据库中检索得到的(如上述子步骤222中所述)。
85.接下来,在子步骤252,基于子步骤251获取的该断点末端列表中的所有断点末端的所有测序序列和前一循环获取的该断点末端列表的局部组装序列集合构建输入序列集
合,并且以大小为第一值k、步长为1的滑动窗依次获取该输入序列集合中的碱基片段。在一种实施例中,在初始化时,可以将第一值k设置为17,从而在子步骤252中以大小为17和步长为1的滑动窗获取该输入序列集合中的碱基片段。在这种情况下,由于不存在前一循环,因此“前一循环获取的该断点末端列表的局部组装序列集合”为空,即输入序列集合仅包含该断点末端列表中的所有断点末端的所有测序序列本身。在生物信息学领域,将这样得到的碱基片段称为kmer,其长度为滑动窗的大小,即该第一值k。通过这种方式,可以将较长的测序序列打断为较短的碱基片段以降低计算规模,并且相邻碱基片段之间彼此部分重叠(例如长度为k的相邻碱基片段之间的重叠部分为k
‑
1个碱基)从而能够降低这些短的碱基片段的数据集中的数据冗余性。
86.接下来,在子步骤253,基于子步骤252获取的碱基片段构建德布莱英图(de bruijn graph,dbg)。dbg包括多个第三节点和多个第三边,每个第三节点为一个碱基片段,每个第三边连接相邻的两个碱基片段。如上所述,相邻的两个碱基片段之间至少部分重叠,因此可以根据碱基片段之间的重叠性将相邻的碱基片段通过第三边相连。测序碱基错误通常是随机的,在高深度测序的条件下,可以通过过滤低频次的碱基片段减少dbg的复杂性。
87.图6示出了根据本发明一种实施例的基于碱基片段构建德布莱英图的子步骤253的流程图。其中,根据一个碱基片段在该输入序列集合中的一个输入序列中是独有碱基片段还是非独有碱基片段,该碱基片段在构建dbg时执行不同的处理。这里,独有碱基片段指示对于输入序列集合中的任意一条输入序列来说,在该输入序列中未重复出现的碱基片段,非独有碱基片段指示在该输入序列中重复出现的碱基片段。
88.具体地,如图6中所示,子步骤253可以进一步包括:子步骤2531,其中确定一个碱基片段是否存在于dbg中。如果确定该碱基片段不存在于dbg中(子步骤2531中判断为“否”),在子步骤2532,将该碱基片段作为一个第三节点添加到dbg中,并且在该碱基片段和该碱基片段的前一碱基片段之间添加第三边。
89.另一方面,如果确定该碱基片段存在于dbg中(子步骤2531中判断为“是”),在子步骤2533,确定该碱基片段是独有碱基片段还是非独有碱基片段。
90.如果在子步骤2533确定该碱基片段是独有碱基片段,则在子步骤2534,更改该独有碱基片段与该独有碱基片段的前一碱基片段之间的第三边的权重。
91.另一方面,如果在子步骤2533确定该碱基片段是非独有碱基片段,则在子步骤2535,获取该非独有碱基片段与该非独有碱基片段的前一碱基片段之间的第三边,并且在子步骤2536,确定是否获取到该非独有碱基片段与该非独有碱基片段的前一碱基片段之间的第三边。
92.如果在子步骤2536获取到该非独有碱基片段与该非独有碱基片段的前一碱基片段之间的第三边,则在子步骤2537,更改该非独有碱基片段与该非独有碱基片段的前一碱基片段之间的第三边的权重。
93.另一方面,在子步骤2536没有获取到该非独有碱基片段与该非独有碱基片段的前一碱基片段之间的第三边,则在子步骤2538,将该非独有碱基片段作为一个第三节点添加到dbg中,并且在该非独有碱基片段和该非独有碱基片段的前一碱基片段之间添加第三边。
94.构建dbg的伪代码可以如下所示:procedure addnode(kmer, pre_kmer, dbg)
if iskmeruniq(kmer) then
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
//独有kmer片段
ꢀꢀꢀꢀ
if kmer not in dbg then
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
//不在dbg中
ꢀꢀꢀꢀꢀꢀꢀꢀ
addnodebase(kmer, dbg)
ꢀꢀꢀꢀꢀꢀꢀꢀ
addedgebase(kmer, pre_kmer, dbg)
ꢀꢀꢀꢀ
else
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
//已经在dbg中
ꢀꢀꢀꢀꢀꢀꢀꢀ
addedgebase(kmer, pre_kmer, dbg)else
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
//非独有kmer片段
ꢀꢀꢀꢀ
if kmer not in dbg then
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
//不在dbg中
ꢀꢀꢀꢀꢀꢀꢀꢀ
addnodebase(kmer, dbg)
ꢀꢀꢀꢀꢀꢀꢀꢀ
addedgebase(kmer, pre_kmer, dbg)
ꢀꢀꢀꢀ
else
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
//在dbg中
ꢀꢀꢀꢀꢀꢀꢀꢀ
edge <
‑ꢀ
getedge(kmer, pre_kmer, dbg)
ꢀꢀꢀꢀꢀꢀꢀꢀ
//取边
ꢀꢀꢀꢀꢀꢀꢀꢀ
ifedge then
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
edge.weight <
‑ꢀ
edge.weight + w
ꢀꢀꢀꢀ
//每次增加权重w
ꢀꢀꢀꢀꢀꢀꢀꢀ
else
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
addnodebase(kmer, dbg)addedgebase(kmer, pre_kmer, dbg)注意,出于编程模块化考虑,这里的伪代码的处理逻辑与上述图6中所示的子步骤2531至2538并不完全一致,但是其包含的思想是一致的。
95.根据上述图6所示的子步骤获取的dbg可能并不是优化的dbg,例如该dbg中可能存在环路(loop)、泡状结构(bubble)、弱末梢(weak tips)等,也可以将其称为初始dbg。在子步骤253的进一步处理中,可以对该初始dbg进修剪以得到更加精简的dbg。
96.具体地,可以对该初始dbg执行以下操作中的至少一种来构建精简的dbg。
97.1)检测该初始dbg以确定是否存在环路,如果存在环路,则跳过本次循环。例如,直接转入子步骤255以将k值增加预定值。
98.2)检测该初始dbg以确定是否存在泡状结构,其中泡状结构具有两条路径,这两条路径具有相同的开始第三节点和结束第三节点。
99.如果确定存在泡状结构,则基于这两条路径上的第三节点的数量和第三边的权重从该初始dbg中去除这两条路径中的一条路径。具体地,可以计算每条路径的权重之和,并且去除权重之和较低的一条路径。
100.3)检测该初始dbg以确定是否存在弱末梢,其中弱末梢表示每个第三节点数目小于第一阈值(例如1000)并且每个第三边的权值低于第二阈值(例如2)的末端节点自由(末端节点自由是指末端节点入度为0或末端节点出度为0)路径。
101.如果确定存在这种弱末梢,则从初始dbg中去除该弱末梢。由于旧的弱末梢去除后可能产生新的弱末梢,弱末梢去除的过程是迭代的,重复地去除dbg中的弱末梢,直到所有弱末梢都被去除,或者达到最大迭代次数(例如10次)。
102.4)检测初始德布莱英图以确定是否存在分支状的头和尾,其中头表示从入度为0的节点开始的路径,尾表示以出度为0的节点结束的路径,分支状的头表示连接到同一节点的数目至少为2的头的集合,分支状的尾表示连接到同一节点的数目至少为2的尾的集合。
103.如果确定存在分支状的头,则合并初始德布莱英图中的分支状头。如果确定存在分支状的尾,则合并初始德布莱英图中的分支状的尾。合并的方法为,获取分支状头集合中权重之和最大的路径,获取每个头路径的碱基序列,计算其他头序列与权重最大的头序列之间的海明距离,如果海明距离小于预设值(例如5),那么将该头路径合并到权重最大头路径中,否则删除该头路径。分支状尾的处理方法与分支状头一致。
104.通过这种方式,可以对根据图6的方式所构建的初始dbg进行修剪(pruning),以获得更加优化的dbg以便于后续获取局部组装序列集合。
105.继续图5,在子步骤254,可以基于dbg中的多个第三节点的碱基片段之间的重叠关系获取该dbg的局部组装序列集合。
106.如前所述,dbg中相邻第三节点的碱基片段是有重叠的,基于相邻碱基片段之间的重叠关系,遍历dbg中的最大非分支子图(maximal non
‑
branching subgraph),可以组装得到连续的序列,称为局部组装序列(contig)。虽然dbg已经进行了修剪,dbg中仍然可能存在多个最大非分支子图,所以可能产生多个局部组装序列,称为局部组装序列集合c
k
。
107.上述子步骤252至254可以多次重复,每次重复构成一个循环,其中在每个循环中,第一值k可以相对于上一个循环的第一值k增大预定值δ(称为步长)。此外,还可以设置第一值k的上限(称为第二值k
max
),以控制循环次数。例如,如上所述,可以将第一值k的初始值k
min
设置为17,第二值k
max
设置为97,并且将步长δ设置为10,重复执行上述子步骤252至254,以获取新的局部组装序列集合。
108.具体地,如图5中所示,在子步骤255,将第一值k增大该预定值δ,并且在子步骤256,确定增大后的第一值k是否小于或等于第二值k
max
。
109.如果确定增大后的第一值k小于或等于第二值k
max
,流程转至子步骤252,基于该断点末端列表中的所有断点末端的所有测序序列和局部组装序列集合c
k
构建用于下一循环的输入序列集合并且以大小为增大后的第一值k(例如k+10)、步长为1的滑动窗查找该输入序列中的碱基片段。这里,在第一次循环时,输入序列集合可以只包括该断点末端列表中的所有断点末端的所有测序序列,而在第二次和之后的循环中,输入序列集合包括该断点末端列表中的所有断点末端的所有测序序列和上一循环的局部组装序列集合。
110.另一方面,如果确定增大后的第一值k大于第二值k
max
,流程转至子步骤257,输出得到的局部组装序列集合。即,子步骤257的最终输出结果是最后一个循环得到的局部组装序列集合。
111.步骤250中所述的局部组装方法,在构图阶段引入非独有kmer,解决了重复序列组装困难的问题。并且该方法的整体流程使用迭代式方式,将上一次组装生成的局部组装序列集合和断点末端的测序序列一起作为下一次组装的输入序列集合,能够同时利用小kmer的组装优势(有测序错误时组装比大kmer好、低覆盖度区域组装比大kmer好)和大kmer的组装优势(重复序列组装比小kmer好)。对二代测序技术而言,插入变异的检测难度高,非常依赖于序列组装算法。然而,现有技术中实现或使用的组装算法都未针对结构变异证据的特征(重复序列、覆盖不均匀等)进行优化,因此检测插入变异的敏感度低。例如,manta使用隐式实现的dbg算法进行局部组装,svaba使用基于string graph的第三方软件库sga进行局部组装,gridss使用colored dbg进行局部组装。
112.获取的局部组装序列集合可能为空。例如,如上所述,如果碱基片段长度k等于k
max
时dbg中仍然有环,或者由于参与组装的序列数目太少,去除弱末梢时删除了所有的第三节点,则此时可能获得的局部组装序列集合为空。
113.如果获取的局部组装序列集合不为空,可以将局部组装序列集合中的每条局部组装序列与参考基因组进行比对,通过比对结果检测该断点末端列表的第二候选结构变异集合。如果局部组装序列集合为空,那么第二候选结构变异集合为空。这种比对可以通过minimap2或bwa
‑
mem软件库来实现。
114.继续图2,接下来,在步骤260,设备100(例如候选结构变异集合确定单元125)确定步骤240获取的第一候选结构变异是否包含于步骤250获取的第二候选结构变异集合,并且如果第一候选结构变异包含于第二候选结构变异集合,那么将第二候选结构变异集合确定为该断点末端列表的候选结构变异集合。如果第一候选结构变异不包含于第二候选结构变异集合,那么在步骤270,将第一候选结构变异和第二候选结构变异集合的并集确定为该断点末端列表的候选结构变异集合。
115.通过这种方式,对于能够通过局部组装和比对方式获得的候选结构变异,可以通过推测的第一候选结构变异和局部组装得到的第二候选结构变异集合一起确定该断点末端列表的候选结构变异集合。而对于不能通过局部组装和比对方式获得的候选结构变异集合(例如由于序列重复、测序错误、测序覆盖度低等原因),则可以以该推测的第一候选结构变异作为该断点末端列表的候选结构变异集合,可以提升结构变异检测的敏感性。
116.继续图2,在步骤280,设备100(例如结构变异确定单元126)基于步骤260或270获得的所有断点末端列表的所有候选结构变异集合确定基因组结构变异。
117.如前所述,构建了一个全基因组或目标基因区间的断点末端区间图;对于断点末端区间图中的每条边,构建了一个断点末端图;对于每个断点末端图,可以得到一个或多个独立子图;对于每个独立子图,可以聚类产生一个或多个断点末端列表;对于每个断点末端列表,可以确定相应的候选结构变异集合。因此,步骤260和270所获得的候选结构变异集合是整个基因组的局部候选结构变异,在步骤280中,可以首先基于这些局部候选结构变异确定每个断点末端列表的结构变异,然后再根据所有断点末端列表的结构变异确定整个基因组或目标基因区间的结构变异。
118.图7示出了根据本发明一种实施例的用于确定基因组结构变异的步骤280的流程图。
119.本发明使用贝叶斯方法计算候选结构变异的基因型(genotype)和质量值(quality score)。为了简化计算,本发明仅考虑一种替代等位基因结构变异,即等位基因集合a={r, x},其中r为参考等位基因,x为结构变异等位基因。本发明仅考虑二倍体的情况,因此有基因型集合g={rr,rx,xx},rr表示纯合参考,rx表示杂合突变,xx表示纯合突变。如图7中所示,步骤280可以基于每个断点末端列表的候选结构变异集合中的每个候选结构变异的每种基因型的先验概率和在该基因型的条件下出现观测比对数据的条件概率确定该候选结构变异的每种基因型的后验概率。
120.在子步骤281,使用贝叶斯方法,基于每个断点末端列表的候选结构变异集合的每个候选结构变异的每种基因型的先验概率和在该基因型的条件下出现观测比对数据的条件概率确定该候选结构变异的每种基因型的后验概率。例如,基因型g的后验概率可以通过如下公式(1)计算:
(1)其中为后验概率,即在观测数据d的条件下得到基因型g的条件概率。
121.为基因型g的先验概率,即根据先前观测或研究得出的基因型g出现的概率。例如,可以使用固定的先验概率:
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(2)其中。
122.假设每一个比对数据都是独立的,在基因型g的条件下出现观测数据d的条件概率可以通过如下公式(3)计算:
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(3)其中,a为等位基因变异的集合,为在等位基因变异a的条件下出现观测数据d的条件概率。例如,可以通过如下公式(4)来计算:
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(4)其中为双端序列在等位基因型a和插入片段分布i的条件下的概率,插入片段的长度分布i例如可以如下面步骤210中所述计算得到,i
d
为双端序列的插入片段长度。和分别为测序序列1和测序序列2发生
‘
切分’比对的概率。具体地,的计算方法如下:即,分为两种情况,当双端序列的插入片段长度大于插入片段分布i的中值i
m
时,等位基因a为r时是p
acc
(在插入片段分布i中,从最大插入片段长度i
max
到数据插入片段长度i
d
的累计概率),等位基因a为x时是1减去p
acc
;当双端序列的插入片段长度小于或等于插入片段分布i的中值i
m
时,等位基因a为r时为1减去p
pe
(p
pe
是在发生结构变异的条件下插入片段长度正常的概率,预设值10
‑8),等位基因a为x时为p
pe
。
123.发生
‘
切分’比对的概率的计算方法如下:其中,为序列d
i
比对到包含等位基因a的序列(等位基因a为r时,该序列为局部
参考序列,等位基因a为x时,该序列为局部组装序列)的比对分数,为序列d
i
比对到不包含等位基因a的序列的比对分数,为比对分值差异阈值,预设值例如可以为8,为设定的发生切分比对的错误概率,预设值例如可以为10
‑3。
124.接下来,在子步骤282,可以基于该候选结构变异的每种基因型g的后验概率确定该候选结构变异的每种基因型g的似然性分值(phred
‑
scaled likelihood)。
125.在一种实施例中,可以根据如下公式(5)确定基因型g的似然性分值pl(phred
‑
scaled likelihood round to closest integer):
ꢀꢀ
(5)其中似然性分值pl是如上计算的pl的四舍五入的整值。接下来对pl值进行均一化,所有基因型的pl至都减去pl值的最小值。
126.接下来,计算候选结构变异的基因型和质量值q,结构变异的基因型为pl值为0的基因型,质量值q为纯合参考基因型(rr)的pl值。
127.接下来,在子步骤283,可以基于该候选结构变异的每种基因型g的似然性分值pl,将似然性分值最小的基因型确定为该候选结构变异的基因型。
128.在子步骤284,可以基于该候选结构变异的每种基因型g的似然性分值pl,将纯合参考基因型的似然性分值确定为该候选结构变异的质量值q。
129.此外,这里还可以计算条件基因型质量值gq,条件基因型质量值gq是pl值中按照大小排名,从小到大第二的值。
130.接下来,在子步骤285,确定该候选结构变异的质量值q是否大于预设值。在一种实例中,该预定值可以设置为20。
131.如果确定该候选结构变异的质量值q大于该预定值,则在子步骤286,将该候选结构变异作为该断点末端列表的结构变异。
132.最后,在子步骤287,合并所有断点末端列表的结构变异以获取该基因组的结构变异。
133.该基因组的结构变异例如可以以标准的vcf(variant call format,变异检测格式)输出。
134.在步骤220构建断点末端区间图时,需要基于待检测的基因组的插入片段的长度分布确定测序序列的结构变异信号,或者,在步骤280确定基因组结构变异时需要基于待检测的基因组的插入片段的长度分布确定在等位基因变异a的条件下出现观测数据d的条件概率。
135.这里,待检测的基因组的插入片段的长度分布可以单独确定并预先存储在设备100中,也可以作为方法200的一部分。在作为方法200的一部分实现的情况下,如图2中所示,方法200还可以包括步骤210,其中设备100(如插入片段长度分布确定单元110)可以确定该基因组的插入片段的长度分布。注意,在图2中,虽然将步骤210显示为位于步骤220至280之前,但是本领域技术人员可以理解,步骤210可以位于任意适当的位置。
136.图8示出了根据本发明一种实施例的用于确定基因组的插入片段的长度分布的步骤210的流程图。
137.如图8中所示,步骤210可以包括子步骤211,其中获取待检测的基因组的标准比对文件。如前所述,该标准比对文件包括该基因组的测序序列与该基因组的参考基因组之间
的比对记录。
138.在子步骤212,对参考基因组的每条染色体,不重复地随机抽取第一数量个染色体区间,其中每个染色体区间大小相同并且互不重叠。例如,可以预先设置染色体区间大小(如200kb(千碱基)),并根据该区间大小将该染色体均匀地划分为多个染色体区间。所抽取的染色体区间的数量(第一数量)例如可以为10个。
139.接下来,在子步骤213,对于第一数量个染色体区间中的每个染色体区间,分别从该标准比对文件中提取与该染色体区间重叠的比对记录,并且基于该比对记录确定插入片段。这里,与染色体区间重叠是指比对记录与该染色体区间至少有一个位置重叠。例如,在bam文件中,插入序列的长度直接记录在获取的标准比对文件的每条比对记录中。
140.在子步骤214,统计该第一数量个染色体区间的插入片段的集合的中值和标准差。
141.集合的标准差σ
i
可以通过如下公式(6)和公式(7)计算得到:,
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(6),
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(7)其中,μ
i
表示集合中的m个插入片段的均值,z
j
表示集合中的m个插入片段中的第j个插入片段的长度(j=1, 2,
ꢀ……
m),i表示循环次数。
142.上述子步骤212至214可以作为一个循环重复执行,对于第i个循环,插入片段的集合的中值表示为m
i
,标准差表示为σ
i
。
143.在子步骤215,确定是否满足预定中止条件。这里,预定中止条件可以是预定收敛阈值或是预定循环次数,满足其中的一个即中止迭代。
144.在一种实施例中,可以确定该第一数量个染色体区间的插入片段的集合的中值m
i
和标准差σ
i
的改变量δm(δm=m
i
‑
m
i
‑1)和δσ(δσ=σ
i
‑
σ
i
‑1),并且确定该改变量δm和δσ是否小于预定收敛阈值δm
th
和δσ
th
。如果确定该改变量δm和δσ小于该预定收敛阈值δm
th
和δσ
th
,确定满足该预定中止条件。为子步骤212至214的循环设置计数器。在子步骤214之后,将计数值加一,并且确定该计数值是否大于预定循环次数(例如5)。如果该计数值大于预定循环次数,确定满足该预定中止条件。
145.如果确定满足该预定中止条件(子步骤215的判断为“是”),基于插入片段的集合中的插入片段的长度确定该基因组的插入片段的长度分布。即,使用满足中止条件时的中值和标准差作为本次测序序列数据集合的插入片段的长度分布的中值和标准差。
146.如果确定不满足该预定中止条件(子步骤215的判断为“否”),步骤210进入下一个循环,再次执行子步骤212至子步骤214。注意,在每个循环中,在子步骤212中随机抽取的第一数量个染色体区间应该不同于之前的循环中抽取的染色体区间。
147.图9示出了适合实现本公开的实施例的计算设备900的结构方框图。计算设备900例如可以是如上所述的设备100。
148.如图9中所示,计算设备900可以包括一个或多个中央处理单元(cpu)910(图中仅示意性地示出了一个),其可以根据存储在只读存储器(rom)920中的计算机程序指令或者从存储单元980加载到随机访问存储器(ram)930中的计算机程序指令,来执行各种适当的动作和处理。在ram 930中,还可存储计算设备900操作所需的各种程序和数据。cpu 910、
rom 920以及ram 930通过总线940彼此相连。输入/输出(i/o)接口950也连接至总线940。
149.计算设备900中的多个部件连接至i/o接口950,包括:输入单元960,例如键盘、鼠标等;输出单元970,例如各种类型的显示器、扬声器等;存储单元980,例如磁盘、光盘等;以及通信单元990,例如网卡、调制解调器、无线通信收发机等。通信单元990允许计算设备900通过诸如因特网的计算机网络和/或各种电信网络与其他设备交换信息/数据。
150.上文所描述的方法200例如可由计算设备900(如设备100)的cpu 910执行。例如,在一些实施例中,方法200可被实现为计算机软件程序,其被有形地包括于机器可读介质,例如存储单元980。在一些实施例中,计算机程序的部分或者全部可以经由rom 920和/或通信单元990而被载入和/或安装到计算设备900上。当计算机程序被加载到ram 930并由cpu 910执行时,可以执行上文描述的方法200的一个或多个操作。此外,通信单元990可以支持有线或无线通信功能。
151.本领域技术人员可以理解,图9所示的计算设备900仅是示意性的。在一些实施例中,设备100可以包含比计算设备900更多或更少的部件。
152.实验结果:使用hg002人类结构变异标准数据集进行测试。本发明对所有结构变异的f1值为48.68%,优于manta的46.08%;本发明对插入变异(ins)的f1值为39.48%,优于manta的28.36%;本发明对插入变异的recall为24.79%,优于manta的16.61%,提升幅度高达49.22%。本发明对插入变异的precision与manta相当(低0.01%)。详细结果如下:检测结果中与标准集匹配的结构变异数目为真阳性数目(tp),与标准集不匹配的结构变异数目为假阳性数目(fp),标准集中存在的而检测结果中不存在的结构变异数目为假阴性数目(fn)。召回率(recall),精确度(precision),f1值(召回率和精确度的调和平均值,常用于评估整体检测性能)的计算方法如下:,,。
153.同时,与现有技术相比,本发明的方案可以检出更长的插入变异。例如,长度为200bp以上的插入变异,本发明检出692个,而manta仅检出215个;长度为900bp以上的插入变异,本发明检出37个,而manta检出0个。
154.本发明可以是方法、装置、系统和/或计算机程序产品。计算机程序产品可以包括计算机可读存储介质,其上载有用于执行本发明的各个方面的计算机可读程序指令。
155.在一个或多个示例性设计中,可以用硬件、软件、固件或它们的任意组合来实现本
发明所述的功能。例如,如果用软件来实现,则可以将所述功能作为一个或多个指令或代码存储在计算机可读介质上,或者作为计算机可读介质上的一个或多个指令或代码来传输。
156.本文公开的装置的各个单元可以使用分立硬件组件来实现,也可以集成地实现在一个硬件组件,如处理器上。例如,可以用通用处理器、数字信号处理器(dsp)、专用集成电路(asic)、现场可编程门阵列(fpga)或其它可编程逻辑器件、分立门或者晶体管逻辑、分立硬件组件或用于执行本文所述的功能的任意组合来实现或执行结合本发明所描述的各种示例性的逻辑块、模块和电路。
157.本领域普通技术人员还应当理解,结合本发明的实施例描述的各种示例性的逻辑块、模块、电路和算法步骤可以实现成电子硬件、计算机软件或二者的组合。
158.本发明的以上描述用于使本领域的任何普通技术人员能够实现或使用本发明。对于本领域普通技术人员来说,本发明的各种修改都是显而易见的,并且本文定义的一般性原理也可以在不脱离本发明的精神和保护范围的情况下应用于其它变形。因此,本发明并不限于本文所述的实例和设计,而是与本文公开的原理和新颖性特性的最广范围相一致。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1