一种基于多基因风险打分构建疾病分类模型的方法与流程
本发明属于生物信息学领域,具体涉及一种基于多基因风险打分构建疾病分类模型的方法。
背景技术:
复杂疾病是在众多因素共同作用下发生的。在过去二十年里,人们对研究遗传风险因素对人类行为变化的影响越来越感兴趣。全基因组关联研究(gwas)可以确定单核苷酸多态性(snps)与表型性状之间的关联。gwas方法普遍应用于社会科学领域,已经识别了很多常见的复杂疾病相关联的遗传变异。大部分遗传变异对于疾病风险贡献通常很小,效应值or(oddsratios)通常在1.1到1.5之间,不足以直接预测疾病状态。但是它们却对于疾病的风险有着更强的累积的临床显著效应。
多基因风险打分(prs)是一种统计学的方法,可以根据个体的基因型谱来评估某一疾病或者性状的遗传风险,也就是多个风险位点的累积效应。经典的prs方法中,prs就是个体所有的风险等位以风险等位的效应值加权的总和来计算的。研究发现相对于只采用达到gwas显著性的snp来说,利用大量的snp来计算prs对于疾病状态的预测能力更佳,但由于并不是所有位点都会影响所研究的性状,因此如何找到复杂疾病风险的最佳预测prs,已经成为目前亟待解决的重要问题。
技术实现要素:
本发明的目的是早期发现目标疾病,具体需要构建疾病分类模型实现。
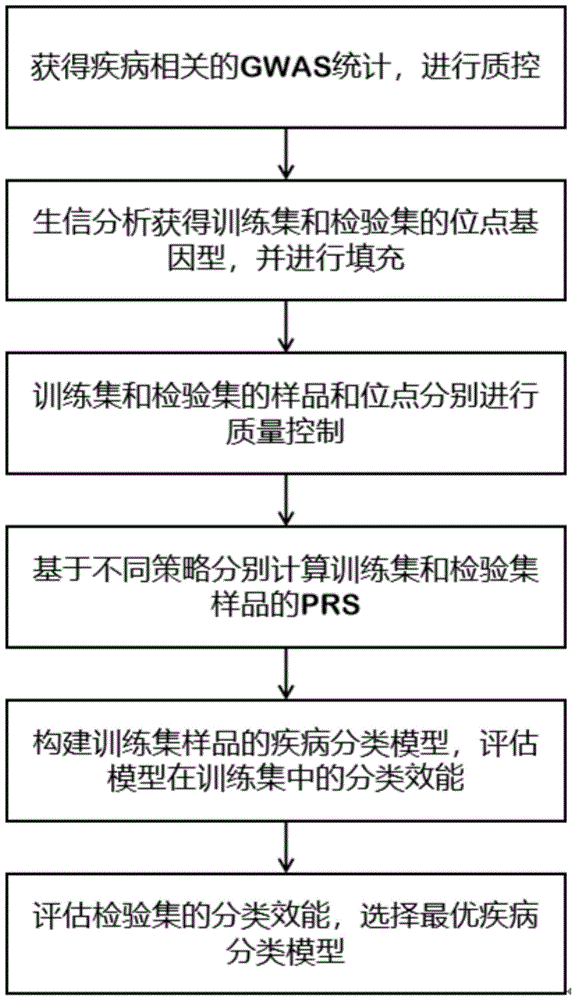
本发明首先保护一种基于多基因风险打分构建疾病分类模型的方法,包括如下步骤:
(1)获得目标疾病的gwas统计数据;gwas统计数据包括位点rs号及其主要等位、次要等位、次要等位的位点效应值、与疾病关联的显著性p值,对gwas统计数据进行位点的质量控制;
(2)基于snp芯片或测序(如高通量测序)数据获取训练集和检验集的全基因组位点的分型信息(即位点基因型);训练集和检验集均包含疾病及对照样品;
(3)基于千人基因组的位点信息对snp芯片或者测序数据得到的样品的位点信息进行位点填充,获得分析位点的基因型信息;对训练集和检验集分别进行样品质控和位点质控;
(4)基于训练集质控后的样品的质控后的位点和质控后的gwas统计数据,采用不同策略方法中的不同参数分别计算训练集样品的prs;
(5)基于训练集数据,以prs作为自变量,以疾病性状作为因变量构建logistic模型,基于五倍交叉验证获得模型的auc曲线下面积来评估模型在训练集中的分类效能;对多种方法不同参数下计算出的prs分别构建logistic模型;
(6)将训练集计算prs的方法应用于检验集数据,计算样品的prs后获取检验集的模型分类效能auc;筛选训练集评估效能和检验集分类效能最优的模型,该模型即为疾病分类模型。
所述步骤(1)中,获得目标疾病的gwas统计数据可以从数据库下载,也可以从大量数据中分析获取。
所述步骤(1)中,位点效应值可为or或beta值。
所述步骤(1)中,对gwas统计数据进行位点的质量控制可包含去除重复的位点、去除不明确的位点、保留最小等位频率maf大于0.01且填充info值大于0.5或0.8的位点。
所述步骤(1)中,所述不明确位点指参考碱基和变异碱基同时为嘌呤或嘧啶。
所述步骤(2)中,snp芯片或测序数据需经生物信息分析手段获得全基因组范围内的位点及其基因型信息。检验集的疾病病种与训练集相同且检验集与训练集不能包含相同的疾病或正常样品。
所述步骤(3)中,获得分析位点的基因型信息的方法可为基于全基因组范围的位点基因型及千人基因组的数据依次进行定相、位点填充、保留填充位点。合并填充位点及全基因组范围内的位点,即为分析位点。
步骤(3)中,位点填充基于全基因组范围的位点基因型及千人基因组的数据,采用shapeit软件进行定相,采用impute2软件进行位点填充,保留填充info值大于0.5或0.8的填充位点。
所述步骤(3)中,对训练集和检验集分别进行样品质控和位点质控可包括去除重复位点、去除与gwas统计数据等位不同的位点、去除maf值小于0.01的位点、去除样品缺失率大于0.01的位点、去除哈代温伯格频率大于0.000001的位点、去除亲缘关系密切的样品和去除训练集和检验集中的重复样品中的至少一个。
所述步骤(3)中,对分析位点进行质控,可包括去除重复位点、去除与gwas统计数据等位不同的位点、去除maf值小于0.01的位点、去除样品缺失率大于0.01的位点和去除哈代温伯格频率大于0.000001的位点中的至少一个。
所述步骤(3)中,对训练集和检验集样品进行质控可包括去除亲缘关系密切的样品、去除训练集和检验集中的重复样品中的至少一个。
所述步骤(4)中,不同策略主要包含两种收缩策略,由于位点效应值的估计具有不确定性,且并非所有位点都会影响所研究的性状,因此对所有位点使用未经调整的效应估计值可能会产生标准误差较高和预测效能较差的prs。为解决这个问题,采用了两种广泛的收缩策略:通过标准或定制的统计技术收缩调整所有位点的效应估计,使用p值或其他筛选阈值作为纳入位点的标准。
所述步骤(4)中,采用不同策略方法可为pruningandthreading、betashrinkage、lassosum、ldpred和深度神经网络。
所述pruningandthreading方法基于gwas统计数据的p值及训练集的连锁r2来筛选位点集合,筛选标准为p值可设置1、0.5、0.05、0.0005、0.000005、0.00000005,连锁r2值可设置0.2、0.4、0.6、0.8,两个筛选标准任意组合来筛选snp集合;采用plink软件的--score参数基于筛选出的位点集合的效应值及位点的基因型来计算样品的prs;plink计算prs的公式如下:
其中si是第i个snp的效应值,gij代表样品j中snpi的效应等位的个数,p是样品的倍性(人通常是2),n代表计算prs所纳入的snp的数目。mj是样品j中非缺失的snp数目。如果样品的snpi缺失,则采用群体的最小等位频率maf乘以倍性来替代gij。
所述betashrinkage方法首先基于gwas统计数据的p值筛选位点集合,p值可设置1、0.5、0.05、0.0005、0.000005、0.00000005。基于高维贝叶斯线性模型及千人基因组的连锁不平衡数据,对筛选后位点集合的效应值进行多次迭代调整,得到最优效应值,使得分类模型达到最优。采用plink软件的--score参数基于筛选后的位点集合的最优效应值及位点的基因型来计算样品的prs。
所述ldpred方法基于贝叶斯方法利用默认阈值ρ来重新计算位点的所述效应值,ρ值可设置1、0.3、0.1、0.03、0.01、0.003、0.001,利用重新计算的效应值基于ldpred软件的自定义的算法来计算样品的prs。
所述lassosum方法基于线性回归模型构建一个惩罚函数来压缩位点的效应值,使得部分位点的效应值为0,得到最优的位点集合并结合千人基因组的连锁不平衡数据计算样品的prs。
所述深度神经网络方法首先基于gwas统计数据的p值筛选位点集合,p值可设置1、0.5、0.05、0.0005、0.000005、0.00000005;对筛选位点的基因型进行0/1/2编码,0和2编码分别代表2个次要等位和2个主要等位的纯合基因型,1编码代表杂合基因型。采用筛选的位点集合的编码值作为神经网络的输入层,将leakyrectifiedlinearunit(relu)激活函数应用于所有隐藏层的,输出层采用sigmoid激活函数,sigmoid函数的输出范围是0到1,将输出层的0-1范围的值作为样品的prs。
所述步骤(5)中,五倍交叉验证可为将训练集的疾病样品和正常样品随机分为5份,以其中四份的数据作为训练集合来构建logistic模型,以另一份数据作为检验集来检验模型的效能,重复100次五倍交叉验证取auc的中值或均值来评估模型在训练集中的分类效能。
所述步骤(5)中,疾病状态进行0/1编码,其中0编码对应正常对照样品,1编码对应疾病样品。
所述步骤(6)中,分类效能最优的模型是指训练集的auc值尽可能高,且检验集的auc值与训练集的auc值尽可能相近。所述疾病分类模型的训练集和检验集的auc值通常大于0.6,且疾病样品和正常样品的多基因风险打分prs存在显著差异。
采用上述任一所述的方法构建的疾病分类模型也属于本发明的保护范围。
本发明还保护上述任一所述疾病分类模型的应用,可为a1)-a4)中的至少一种:
a1)评估待测者目标疾病的风险;
a2)制备用于评估待测者目标疾病的风险的产品;
a3)预防目标疾病;
a4)制备用于预防目标疾病的产品。
本发明公开一种基于多基因风险打分构建疾病分类模型的方法,该方法可以帮助临床早期发现、预防目标疾病,具有重要的应用价值。
附图说明
图1为实施例1开发的多基因风险打分构建疾病分类模型的方法的技术路线图。
图2为实施例2中训练集和检验集的prs分布图。
具体实施方式
下面结合具体实施方式对本发明进行进一步的详细描述,给出的实施例仅为了阐明本发明,而不是为了限制本发明的范围。以下提供的实施例可作为本技术领域普通技术人员进行进一步改进的指南,并不以任何方式构成对本发明的限制。
下述实施例中的实验方法,如无特殊说明,均为常规方法,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
实施例1、基于多基因风险打分构建疾病分类模型的方法的开发
本发明的发明人在gwas统计数据和大量训练集和检验集的全基因组位点的分型数据基础上,开发了一种基于多基因风险打分构建疾病分类模型的方法,具体包括如下步骤:
1、从公共数据库下载或大量数据中分析获取目标疾病的全基因组关联研究(gwas)统计。gwas统计数据至少包含位点rs号及其主要等位、次要等位、次要等位的位点效应值、与疾病关联的显著性p值,对gwas统计数据进行位点的质量控制。
步骤(1)中,位点效应值为or或beta值。对gwas统计数据进行位点的质量控制包含去除重复的位点、去除不明确的位点、保留最小等位频率maf大于0.01且填充info值大于0.5或0.8的位点。
不明确位点是指参考碱基和变异碱基同时为嘌呤(a或t)或嘧啶(g或c)。
2、基于snp芯片或测序(如高通量测序)数据获取训练集和检验集的全基因组位点的分型信息(即位点基因型)。训练集和检验集均包含疾病及对照样品。
步骤(2)中,检验集的疾病病种与训练集相同,但检验集与训练集不能包含相同的疾病或正常样品。
芯片及测序数据需经生物信息分析手段获得全基因组位点及其基因型信息。
3、基于下载的千人基因组的位点信息对snp芯片或者测序数据得到的样品的位点信息进行位点填充,获得分析位点的基因型信息;对训练集和检验集分别进行样品质控和位点质控。
步骤(3)中,位点填充基于全基因组范围的位点基因型及千人基因组的数据,采用shapeit软件进行定相,采用impute2软件进行位点填充,保留填充info值大于0.5或0.8的填充位点;合并填充位点及全基因组范围内的位点,即为分析位点。
对分析位点进行质控,包括去除重复位点、去除与gwas统计数据等位不同的位点、去除maf值小于0.01的位点、去除样品缺失率大于0.01的位点、去除哈代温伯格频率大于0.000001的位点。
对训练集和检验集样品进行质控,包括去除亲缘关系密切的样品、去除训练集和检验集中的重复样品。
4、基于训练集质控后的样品的质控后的位点和质控后的gwas统计数据,采用不同策略方法中的不同参数分别计算训练集样品的prs。
步骤(4)中,不同策略主要包含两种收缩策略,由于位点效应值的估计具有不确定性,且并非所有位点都会影响所研究的性状,因此对所有位点使用未经调整的效应估计值可能会产生标准误差较高和预测效能较差的prs。为解决这个问题,采用了两种广泛的收缩策略:通过标准或定制的统计技术收缩调整所有位点的效应估计,使用p值或其他筛选阈值作为纳入位点的标准。采用此两种策略相关的五种方法分别计算prs,五种方法分别为pruningandthreading、betashrinkage、lassosum、ldpred和深度神经网络。
pruningandthreading方法基于gwas统计数据的p值及训练集的连锁r2来筛选位点集合,筛选标准为p值可设置1、0.5、0.05、0.0005、0.000005、0.00000005,连锁r2值可设置0.2、0.4、0.6、0.8,两个筛选标准任意组合来筛选snp集合;采用plink软件的--score参数基于筛选出的位点集合的效应值及位点的基因型来计算样品的prs;plink计算prs的公式如下:
其中si是第i个snp的效应值,gij代表样品j中snpi的效应等位的个数,p是样品的倍性(人通常是2),n代表计算prs所纳入的snp的数目。mj是样品j中非缺失的snp数目。如果样品的snpi缺失,则采用群体的最小等位频率maf乘以倍性来替代gij。
betashrinkage方法首先基于gwas统计数据的p值筛选位点集合,p值可设置1、0.5、0.05、0.0005、0.000005、0.00000005。基于高维贝叶斯线性模型及千人基因组的连锁不平衡数据,对筛选后位点集合的效应值进行多次迭代调整,得到最优效应值,使得分类模型达到最优。采用plink软件的--score参数基于筛选后的位点集合的最优效应值及位点的基因型来计算样品的prs。
ldpred方法基于贝叶斯方法利用默认阈值ρ来重新计算位点的所述效应值,ρ值可设置1、0.3、0.1、0.03、0.01、0.003、0.001,利用重新计算的效应值基于ldpred软件的自定义的算法来计算样品的prs。
lassosum方法基于线性回归模型构建一个惩罚函数来压缩位点的效应值,使得部分位点的效应值为0,得到最优的位点集合并结合千人基因组的连锁不平衡数据计算样品的prs。
深度神经网络方法首先基于gwas统计数据的p值筛选位点集合,p值可设置1、0.5、0.05、0.0005、0.000005、0.00000005;对筛选位点的基因型进行0/1/2编码,0和2编码分别代表2个次要等位和2个主要等位的纯合基因型,1编码代表杂合基因型。采用筛选的位点集合的编码值作为神经网络的输入层,将leakyrectifiedlinearunit(relu)激活函数应用于所有隐藏层的,输出层采用sigmoid激活函数,sigmoid函数的输出范围是0到1,将输出层的0-1范围的值作为样品的prs。
5、基于训练集数据,以prs作为自变量,以疾病性状作为因变量构建logistic模型,基于五倍交叉验证获得模型的auc曲线下面积来评估模型在训练集中的分类效能。
对多种方法不同参数下计算出的prs分别构建logistic模型。
步骤(5)中,疾病状态进行0/1编码,其中0编码对应正常对照样品,1编码对应疾病样品。五倍交叉验证为将训练集的疾病样品和正常样品随机分为5份,以其中四份的数据作为训练集合来构建logistic模型,以另一份数据作为检验集来检验模型的效能。重复100次五倍交叉验证,取auc的中值或均值来评估模型在训练集中的分类效能。
6、将训练集计算prs的方法应用于检验集数据,计算样品的prs后获取检验集的模型分类效能auc;筛选训练集评估效能和检验集分类效能最优的模型,该模型即为最佳疾病分类模型。
步骤(6)中,评估效能最优是指训练集的auc值尽可能高,且检验集的auc值与训练集的auc值尽可能相近。最佳疾病分类模型的训练集和检验集的auc值通常大于0.6,且疾病样品和正常样品的多基因风险打分prs存在显著差异。
实施例2、采用实施例1建立的方法构建冠心病分类模型
1、从gwascatalog数据库下载185000例冠心病的正常和对照样品的gwas统计数据结果,共计945万个位点的结果。
2、通过illumina的asa芯片实验及生物信息学分析获得1800例训练集样品(包括900例正常和900例对照)和1500例检验集样品(包括760例正常和740例对照)的73.8万个位点的基因型数据。
3、对训练集和检验集样品进行质控。质控后基于千人基因组数据的单倍型组成,根据训练集和检验集样品缺失位点的周围非缺失位点,判断单倍型的类型,然后根据单倍型的基因型对该样品的缺失位点进行填充,控制info大于0.5,得到共计212万个位点基因型数据。
对训练集和检验集样品填充后的基因型数据分别进行质控,去除gwas统计数据中不包含的位点,去除亲缘关系近的样品。
对下载的gwas统计数据进行质控,去除maf小于0.01的位点、去除填充info值小于0.8的位点、去除模糊的snp位点,只保留训练集和检验集均包含的位点,最后剩余130万个位点。
4、将pruningandthreading方法的24种参数、betashrinkage方法的6种参数、lassosum方法、ldpred方法的7种参数及深度神经网络方法的6种参数分别应用于训练集样品,并计算出训练集每个样品的prs。进一步将训练集的prs算法应用于检验集数据,计算出检验集每个样品的prs。
训练集和检验集的prs分布图见图2。
5、以训练集的样品的prs作为自变量,以样品的疾病状态作为因变量构建logistic模型,100次五倍交叉验证计算训练集模型的auc值。同时计算所有检验集的auc值。
6、所有方法中最好的是pruningandthreading算法的p值0.0005和r2值0.2参数,该算法共纳入455个snp位点,其中训练集的auc为0.6211,检验集的auc为0.6205。
由此可见,pruningandthreading算法的p值0.0005和r2值0.2参数为冠心病分类模型可以应用计算普通人的冠心病风险,对临床的早发现和早预防具有重要的应用价值。
以上对本发明进行了详述。对于本领域技术人员来说,在不脱离本发明的宗旨和范围,以及无需进行不必要的实验情况下,可在等同参数、浓度和条件下,在较宽范围内实施本发明。虽然本发明给出了特殊的实施例,应该理解为,可以对本发明作进一步的改进。总之,按本发明的原理,本申请欲包括任何变更、用途或对本发明的改进,包括脱离了本申请中已公开范围,而用本领域已知的常规技术进行的改变。按以下附带的权利要求的范围,可以进行一些基本特征的应用。
- 还没有人留言评论。精彩留言会获得点赞!