一种布洛芬颗粒及其制备方法与流程
本发明属于药物领域,特别涉及一种布洛芬颗粒及其制备方法。
背景技术:
布洛芬为非甾体消炎药,主要是抑制环氧化酶和前列腺素的合成而达到抗炎镇痛作用,并通过下丘脑体温调节中心起到退热作用,具有较大的推广作用,具有较大的推广应用作用。临床上,一般适用于缓解类风湿关节炎、骨关节炎、脊柱关节病、痛风性关节炎、风湿性关节炎等各种疾病。
目前常用的布洛芬制备方法包括制备速释部分,即将布洛芬、崩解剂、粘合剂混合、湿法制粒以及烘干得到速释颗粒;制备缓释部分,将布洛芬、缓蚀剂、崩解剂、粘合剂通过挤出滚圆的方法指的颗粒,然后包衣得到缓释部分,最后将速释部分和缓释部分压制得到布洛芬。
相关技术提供的方法制备的布洛芬药物成分因粒密度差异容易出现离析现象,溶解性能差。
技术实现要素:
为了解决相关技术提供的方法制备的布洛芬药物成分因粒密度差异容易出现离析现象,溶解性能差的技术问题,本发明提供了一种布洛芬颗粒及其制备方法。
本发明具体技术方案如下:
本发明提供了一种布洛芬颗粒,所述布洛芬颗粒包括:
布洛芬0.8-1.2重量份,填充剂3.0-4.0重量份,粘合剂和崩解剂0.10-2重量份,调味剂和矫味剂0.001-0.002重量份。
在一种可选地实施例中,所述布洛芬颗粒包括:布洛芬1重量份,填充剂3.7重量份,粘合剂和崩解剂0.15重量份,调味剂和矫味剂0.001重量份。
在一种可选地实施例中,所述填充剂为乳糖,所述粘合剂为羟丙纤维素,所述崩解剂为交联羧甲纤维素钠,所述调味剂为富马酸,所述矫味剂为糖精钠。
另一方面,本申请实施例还提供了一种布洛芬颗粒制备方法,所述布洛芬颗粒制备方法包括:
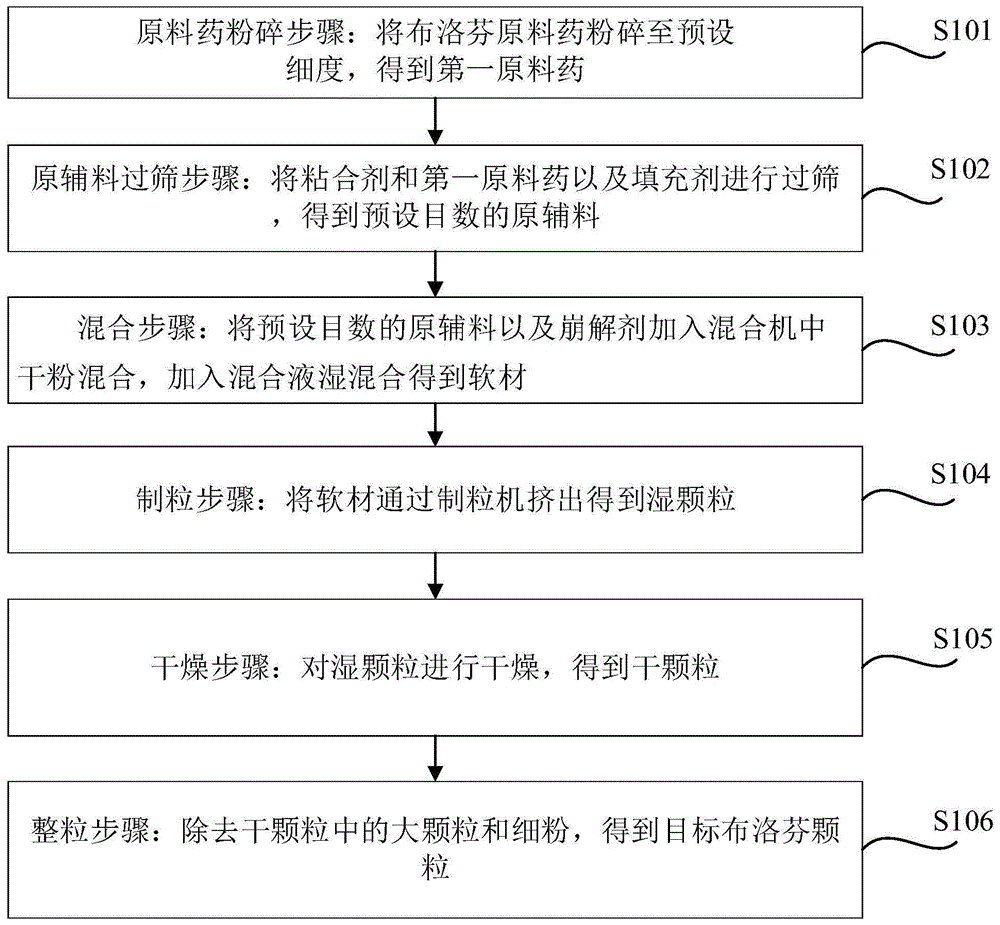
原料药粉碎步骤:将布洛芬原料药粉碎至预设细度,得到第一原料药;
原辅料过筛步骤:将粘合剂和第一原料药以及填充剂进行过筛,得到预设目数的原辅料;
混合步骤:将预设目数的原辅料以及崩解剂加入混合机中干粉混合,加入混合液湿混合得到软材;
制粒步骤:将所述软材通过制粒机挤出得到湿颗粒;
干燥步骤:对所述湿颗粒进行干燥,得到干颗粒;
整粒步骤:除去所述干颗粒中的大颗粒和细粉,得到目标布洛芬颗粒。
在一种可选地实施例中,所述原料药粉碎步骤包括:通过粉碎机在预设电流以及预设频率下以预设加料频率匀速加入所述布洛芬原料粉碎至预设细度。
在一种可选地实施例中,所述预设细度为2μm≤d50≤18μm,4μm≤d90≤56μm。
在一种可选地实施例中,所述预设目数为20目。
在一种可选地实施例中,所述方法还包括配制混合液步骤:将富马酸和矫味剂以及纯化水按1:1:1200比例混合得到所述混合液。
在一种可选地实施例中,所述方法还包括配料混合步骤:将所述预设目数的原辅料以及崩解剂加入混合机中干混;再加入混合液湿混得到软材。
在一种可选的实施例中,所述方法还包括制粒步骤:将软材通过制粒机挤出得到湿颗粒。
在一种可选的实施例中,所述方法还包括干燥步骤:对湿颗粒进行干燥,得到湿颗粒。
在一种可选的实施例中,所述方法还包括整粒步骤:除去干颗粒中的大颗粒和细粉,得到目标布洛芬颗粒。
在一种可选地实施例中,所述方法还包括总混步骤:将所述目标布洛芬颗粒加入混合机中进行整体混合;
优选的,所述方法还包括中间体检测步骤:按照取样规则对所述目标布洛芬颗粒进行取样检测,将符合检测标准的布洛芬颗粒作为所述目标布洛芬颗粒。
本发明的有益效果如下:
本申请实施例提供的布洛芬颗粒,通过上述组分协同复配,使得制备的布洛芬颗粒具有与原研药相同的生物等效性与质量一致性,且制备工艺简单可靠。
附图说明
图1为本申请实施例提供的布洛芬颗粒制备方法流程示意图。
具体实施方式
下面结合附图和以下实施例对本发明作进一步详细说明。
本申请实施例提供了一种布洛芬颗粒,该布洛芬颗粒包括:布洛芬0.8-1.2重量份,填充剂3.0-4.0重量份,粘合剂和崩解剂0.10-2重量份,调味剂和矫味剂0.001-0.002重量份。
本申请实施例提供的布洛芬颗粒,通过上述组分协同复配,使得制备的布洛芬颗粒具有与原研药相同的生物等效性与质量一致性,且制备工艺简单可靠。
以下将通过可选地实施例进一步解释和描述本申请实施例提供的布洛芬颗粒。
在一种可选地实施例中,布洛芬可以为0.8重量份、0.9重量份、1重量份、1.1重量份、1.2重量份等,填充剂3重量份、3.1重量份、3.2重量份、3.3重量份、3.4重量份、3.5重量份、3.6重量份、3.7重量份、3.8重量份、3.9重量份或4重量份等,粘合剂和崩解剂可以为0.1重量份、0.11重量份、0.12重量份、0.13重量份、0.14重量份、0.15重量份、0.16重量份、0.17重量份、0.18重量份、0.19重量份或2重量份等,调味剂和矫味剂可以为0.001重量份、或0.002重量份等。
在一种可选地实施例中,布洛芬颗粒包括:布洛芬1重量份,填充剂3.7重量份,粘合剂和崩解剂0.15重量份,调味剂和矫味剂0.001。
在一种可选地实施例中,填充剂为乳糖,粘合剂为羟丙纤维素,崩解剂为交联羧甲纤维素钠,其中调味剂为富马酸,矫味剂为糖精钠。
另一方面,本申请实施例还提供了一种布洛芬颗粒制备方法,该布洛芬颗粒制备方法包括:
s101、原料药粉碎步骤:将布洛芬原料药粉碎至预设细度,得到第一原料药。
s102、原辅料过筛步骤:将粘合剂和第一原料药以及填充剂进行过筛,得到预设目数的原辅料。
s103、混合步骤:将第一原料药、预设目数的粘合剂、填充剂以及崩解剂加入混合机中干粉混合,加入混合液湿混得到软材。
s104、制粒步骤:将软材通过制粒机挤出得到湿颗粒。
s105、干燥步骤:对湿颗粒进行干燥,得到干颗粒。
s106、整粒步骤:除去干颗粒中的大颗粒和细粉,得到目标布洛芬颗粒。
本申请实施例提供的方法,通过原料药粉碎步骤将布洛芬原料药粉碎至预设细度,将原料药粉中颗粒的细度都粉碎至相同或相近,减少了布洛芬原料药颗粒之间的颗粒密度差异,使布洛芬原料药与原辅料混合时避免出现离析现象;通过原辅料过筛步骤对粘合剂和第一原料药以及填充剂进行过筛,使粘合剂和第一原料药以及填充剂的目数保持一致,减小了原料药与原辅料之间、原料药与原料药之间、原辅料与原辅料之间颗粒间距,提高了原料药与原辅料以及粘合剂之间的结合度,同时将调味剂及矫味剂溶解在纯化水中作为润湿剂,进而提高药物的均一性及溶出性能。
以下将通过可选地实施例进一步解释和描述本申请实施例提供的制备方法。
s101、原料药粉碎步骤:将布洛芬原料药粉碎至预设细度,得到第一原料药。
本申请实施例提供的原料药粉碎步骤中可以通过气流式粉碎机对原料药进行粉碎。
在一种可选地实施例中,原料药粉碎步骤包括:通过粉碎机在预设电流以及预设频率下以预设加料频率匀速加入布洛芬原料粉碎至预设细度。作为一种示例,可以设定气流式粉碎机分级电机电流为2a,分级轮频率为80hz,加料频率为20hz,匀速加入物料,对布洛芬原料药进行粉碎。
在一种可选地实施例中,预设细度为2μm≤d50≤18μm,4μm≤d90≤56μm。
需要说明的是,d50是指一个样品的累计粒度分布百分数达到50%时所对应的粒径。它的物理意义是粒径大于它的颗粒占50%,小于它的颗粒也占50%,d50也叫中位径或中值粒径。d90是指一个样品的累计粒度分布百分数达到90%时所对应的粒径。它的物理意义是粒径小于它的颗粒占90%,大于它的颗粒占10%。
s102、原辅料过筛步骤:将粘合剂和第一原料药以及填充剂进行过筛,得到预设目数的原辅料。
在一种可选地实施例中,预设目数为20目。
在一种可选地实施例中,粘合剂和第一原料药过筛可以采用手工过筛筛网目数可以为20目,填充剂可以采用摇摆式颗粒机过筛、筛网目数可以为20目。
在一种可选地实施例中,该方法还包括配料步骤:将第一原料药与预设目数的粘合剂、填充剂以及崩解剂加入混合机中进行干粉混合。
在一种可选地实施例中,方法还包括配制混合液步骤:将调味剂和矫味剂以及纯化水按1:1:1200比例混合得到混合液。
在一种可选地实施例中,调味剂为富马酸,矫味剂为糖精钠。
进一步地,将富马酸与糖精钠以及纯化水按1:1:1200比例配成溶液,备用。需要说明的是,可以在配制混合液之前用少量40℃~50℃温水溶解富马酸和糖精钠,以提高富马酸和糖精钠溶解速率及混合均匀度。
s103、混合步骤:将预设目数的原辅料以及崩解剂加入混合机中干粉混合,加入混合液湿混合得到软材。
进一步地,将布洛芬、乳糖、羟丙纤维素及交联羧甲纤维素钠加入混合机中,干粉混合40分钟。加入富马酸和糖精钠混合液,湿混约5分钟,制成软材,软材要求手捏能成团,轻压即散,湿润均匀,色泽均匀一致。
s104、制粒步骤:将软材通过制粒机挤出得到湿颗粒。
将制好的软材加入旋转式颗粒机中进行旋转挤出,并及时检查挤出物是否正常。设定球形抛丸机风速20hz~50hz,转盘转速10hz~30hz,加入挤出物10~30kg/次,随时观察物料流化状态,物料成丸状况,使物料在旋转式颗粒机内维持良好的流化状态;完成抛丸作业后出料,出料的湿颗粒转移到高效沸腾干燥机的料斗中,待最后一次出料的物料转移完成后,开始干燥步骤。
s105、干燥步骤:对湿颗粒进行干燥,得到干颗粒。
滚圆后的湿颗粒应投入高效沸腾干燥机的料斗中,设定干燥用空气进风温度60℃,风速30hz~50hz,蒸汽压力0.2mpa~0.4mpa,每5分钟记录一次进风温度、出风温度及物料温度,待物料温度达到50℃以上后,再干燥5分钟后,关闭蒸汽,要求干燥失重不得过1.5%,待物料温度低于40℃后出料。
s106、整粒步骤:除去干颗粒中的大颗粒和细粉,得到目标布洛芬颗粒。
干燥后干颗粒过16/60目三元旋振筛,除去大颗粒和细粉。将整粒后合格的颗粒装入洁净的低密度聚乙烯袋中,扎严,等待总混。
在一种可选地实施例中,方法还包括总混步骤:将目标布洛芬颗粒加入混合机中进行整体混合。
将目标布洛芬颗粒加入三维运动混合机内,转速8转/分,混合10分钟。混合好的颗粒装入洁净的双层低密度聚乙烯袋中,扎严,送入中间站(温度18~26℃,湿度≤65%,中间体颗粒储存时限为28天),等待检测。
在一种可选地实施例中,方法还包括中间体检测步骤:按照取样规则对目标布洛芬颗粒进行取样检测,将符合检测标准的布洛芬颗粒作为目标布洛芬颗粒。
按取样规程进行取样送检,本申请实施例提供的布洛芬颗粒性状为白色颗粒;6个点的含量相对标准偏差rsd应不大于2.0%;本申请实施例提供的布洛芬(按c13h18o2计)应为19.0%~21.0%;干燥失重不得过1.5%;双筛分法测中间体粒度,不能通过1号筛与能通过5号筛的总和不超过供试量的15%;颗粒溶出度(溶出度:指活性药物从片剂、胶囊剂或颗粒剂等普通制剂在规定条件下溶出的速率和程度)15分钟大于等于标示量的85%。
在一种可选地实施例中,方法还包括分装:设定好横封温度(100℃~170℃)、纵封温度(130℃~220℃),压缩空气范围为0.45mpa~0.8mpa,将检验合格的颗粒装入分装机料斗内,调节装量(1g/袋,装量差异±9%),运行速度180~220袋/分。调整轧口处批号,开动机器将颗粒装入塑料膜内。每60分钟检查装量,要求封口严密,不漏气,批号清晰、正确。按外包装工序标准操作规程执行。
以下将通过具体的实施例进一步描述本申请实施例提供的制备方法和制备的布洛芬颗粒。
实施例
(1)原料药粉碎:设定气流式粉碎机分级电机电流2a,分级轮频率80hz,加料频率20hz,匀速加入布洛芬原料药,对布洛芬原料药进行粉碎,粉碎后的布洛芬原料药应至预设细度(布洛芬原料药粒径检测报告:细度为2μm≤d50≤18μm,4μm≤d90≤56μm)。
(2)原辅料过筛:羟丙纤维素及粉碎后的布洛芬原料药用手工筛过筛,手工筛网目数为20目;乳糖用摇摆式颗粒机过筛、筛网目数为20目。
(3)配料:配料前核对原辅料的名称、编号和重量,按批投料量置于双层低密度聚乙烯袋内。
(4)富马酸和糖精钠混合液配制:富马酸、糖精钠、纯化水按1:1:1200比例配成溶液,备用(用少量40℃~50℃温水溶解富马酸和糖精钠)。
(5)混合:将乳糖、布洛芬、羟丙纤维素及交联羧甲纤维素钠加入混合机中,干粉混合40分钟。加入富马酸和糖精钠混合液,湿混约5分钟,制成软材,软材要求手捏能成团,轻压即散,湿润均匀,色泽均匀一致。
(6)制粒:将制好的软材加入旋转式颗粒机中进行旋转挤出,并及时检查挤出物是否正常。设定球形抛丸机风速20hz~50hz,转盘转速10hz~30hz,加入挤出物10~30kg/次,随时观察物料流化状态,物料成丸状况,使物料在容器内维持良好的流化状态;完成抛丸作业后出料,出料的湿颗粒转移到高效沸腾干燥机的料斗中,待最后一次出料的物料转移完成后,开始干燥步骤。
(7)干燥:滚圆后的湿颗粒应投入高效沸腾干燥机的料斗中,设定干燥用空气进风温度60℃,风速30hz~50hz,蒸汽压力0.2mpa~0.4mpa,每5分钟记录一次进风温度、出风温度及物料温度,待物料温度达到50℃以上后,再干燥5分钟后,关闭蒸汽,要求干燥失重不得过1.5%,待物料温度低于40℃后出料。
(8)整粒:干燥后颗粒过16/60目三元旋振筛,除去大颗粒和细粉。将整粒后合格的颗粒装入洁净的低密度聚乙烯袋中,扎严,等待总混。
(9)总混:将颗粒加入三维运动混合机内,转速8转/分,混合10分钟。混合好的颗粒装入洁净的双层低密度聚乙烯袋中,扎严,送入中间站(温度18~26℃,湿度≤65%,中间体颗粒储存时限为28天),待检。
(10)中间体检测:按取样规程进行取样送检,本品性状为白色颗粒;6个点的含量rsd(相对标准偏差)应不大于2.0%;本品含布洛芬(按c13h18o2计)应为19.0%~21.0%;干燥失重不得超过1.5%;双筛分法测中间体粒度,不能通过1号筛与能通过5号筛的总和不超过供试量的15%;颗粒溶出度(溶出度:指活性药物从片剂、胶囊剂或颗粒剂等普通制剂在规定条件下溶出的速率和程度)15分钟大于等于标示量的85%。
(11)分装:设定好横封温度(100℃~170℃)、纵封温度(130℃~220℃),压缩空气范围0.45mpa~0.8mpa,将检验合格的颗粒装入分装机料斗内,调节装量(1g/袋,装量差异±9%),运行速度180~220袋/分。调整轧口处批号,开动机器将颗粒装入塑料膜内。每60分钟检查装量,要求封口严密,不漏气,批号清晰、正确。
(12)外包装:按外包装工序标准操作规程执行。(13)入库。
本申请对采用上述方法制备的布洛芬颗粒进行了评估,并出具了评估报告,经过评估证明本申请实施例提供的方法制备的布洛芬颗粒具有很好的药物效果,并且不会出现离析现象,溶解性能好。
本申请实施例提供的方法制备的布洛芬颗粒与原研药品等效,质量均一。如采用其他方法制备,例如布洛芬不经过粉碎、过筛,原辅料不经过过筛,会导致原辅料混合不均匀,最终制得的颗粒含量不均一,不符合质量标准的要求;或者改变混合液的比例,制得的软材润湿度不够或者过于润湿,在制粒过程中会导致粉状或者黏度过大不成颗粒。
本申请实施例进行评估的处方组成如下所示:
表1:布洛芬颗粒处方组成对比
表1中,本申请实施例提供的制剂布洛芬颗粒(0.2g)处方以及原辅料用量均符合fda批准的药物非活性成分数据库列出的用量,本申请实施例中为更严格控制非活性成分最大使用量,选择已批准剂型中最大使用量中,规定的数值最小的一个,即富马酸的用量选择胶囊剂项下的规定用量0.7mg。
表1中,黏合剂羟丙纤维素用量小于3%时,挤出干燥后所得颗粒粉末较多,黏合剂用量高于3%时,软材旋转挤出后得到的挤出物呈长条状,黏性较大,抛丸滚圆过程中容易黏连成大团块,以上两者均会在整粒工序筛除,废品率增大。
表1中,崩解剂交联羧甲纤维素钠的用量小于3%时,颗粒中活性药物布洛芬的溶出速率和和程度会降低,会影响最终颗粒的溶出度。
采用本申请实施例提供的方法连续生产的工艺验证批次样品(批号:34180601、34180602、34180603)与参比制剂(批号:k64670)进行了全面的质量对比研究,对比结果说明本申请提供制剂与参比制剂质量一致。
检验结果汇总表表2
需要说明的是,表2中的c-022-01、c-022-02、c-022-03、c-022-04、c-022-05、c-022-06、c-022-07、c-022-08、c-022-010为(ibuprofen)杂质。具体的结构式、分子式及分子量见表3。
表3
从表2可以看出,对比结果说明本申请提供制剂与参比制剂质量一致。
在影响因素试验中,本申请实施例提供制剂布洛芬颗粒分别在高温(50℃)、高湿(rh75%,25℃)、光照(5000lx)条件下去包装放置5、10天,样品的性状、溶出度、含量、有关物质、干燥失重等质量指标均符合规定。加速试验中,本申请提供制剂布洛芬颗粒按市售包装,在温度40℃±2℃、rh75%±5%条件下,放置6个月,三批样品的性状、溶出度、含量、有关物质、干燥失重等质量指标均符合规定。长期试验中,本申请提供制剂布洛芬颗粒按市售包装,在温度30℃±2℃、rh65%±10%条件下,放置36个月,现已进行完34个月,目前试验结果表明三批样品的性状、溶出度、含量、有关物质和干燥失重等质量指标均符合规定。
本申请实施例对采用上述方法制备的布洛芬颗粒进行了随机、开放、两制剂、双周期、交叉空腹及餐后单次给药后健康人体生物等效性试验。
本次空腹试验共筛选62例健康受试者,入组24例,所有受试者完成全部两周期的试验,个体血药浓度数据采集完整,纳入药代动力学和生物等效性计算与统计分析。zk05号受试者有一个采血时间超窗,其余受试者采血时间点均符合计划采集时间点或在方案规定的时间窗内,采用实际采集时间点进行pk参数计算。
24名健康受试者在空腹状态下口服布洛芬颗粒200mg(受试制剂或参比制剂)后,血浆中布洛芬的cmax(药物的峰值浓度)及auc0-t、auc0-∞,其中,auc0为药学研究血药浓度在到达一定时间点的表示,0-t是指在一定时间段内的表示,区别于0-∞也就是无穷远的血药浓度标示。经对数转换后几何均值的比值分别为:111.41%、100.96%和100.99%。其对应的90%置信区间分别为104.98%~118.24%、97.85%~104.17%和97.88%~104.19%,均在80.00~125.00%范围内,符合生物等效性判断标准。即本申请实施例提供的布洛芬颗粒(200mg)与参比制剂ibuprofengranules(
本次餐后试验共筛选66例健康男性受试者,入组38例。38例受试者个体血药浓度数据采集完整,纳入安全性数据分析集和pk分析集。采血时间点除zc20受试者服药后1h采血点超窗,其他均符合计划采集时间点或在方案规定的时间窗内,均按照实际采集时间点进行pk参数计算。
38名健康受试者在餐后状态下口服布洛芬颗粒200mg(受试制剂或参比制剂)后,血浆中布洛芬颗粒的cmax及auc0-t、auc0-∞经对数转换后几何均值的比值分别为:105.40%和100.13%、100.18%,其对应的90%置信区间分别为97.31%-114.17%和97.71%-102.60%、97.75%-102.66%,均在80.00~125.00%范围内,符合生物等效性判断标准。即本申请实施例提供的布洛芬颗粒(200mg)与参比制剂ibuprofengranules(规格:20%1g,商品名:
试验结果表明,采用本申请实施例提供的方法生产的布洛芬颗粒与参比制剂在空腹及餐后给药时均具有生物等效性。
再评价本申请实施例提供的制剂布洛芬颗粒(0.2g)在研发、生产过程、稳定性考察等均符合科学性、完整性、一致性和真实性要求;用于生物等效性研究样品(批号:34180601)是在已取得《药品生产质量管理规范》的口服固体制剂车间生产所得。
以上实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!