一种基于HPV整合的肿瘤新抗原预测方法与流程
一种基于hpv整合的肿瘤新抗原预测方法
技术领域
1.本发明属于生物信息学技术领域,具体涉及一种基于人类基因组中的hpv插入片段预测新抗原的方法。
背景技术:
2.hpv(human papillomavirus)也叫人乳头瘤病毒,是一种dna病毒,属于乳头瘤病毒科乳头瘤病毒属,可以感染人体表皮与粘膜组织。目前发现的hpv病毒有一百多种亚型,其中80多种跟人体密切相关。研究证实hpv感染是宫颈癌的致病因素,绝大多数宫颈癌是由高危型hpv持续感染引起,其中最具代表性的高危型hpv病毒亚型为hpv16和hpv18。从感染hpv到最终发展为宫颈癌大约需要15年的时间。在hpv感染早期,hpv基因组以游离形式存在人染色体外,在该阶段的hpv容易被免疫系统清除。随着癌前病变的进展,hpv基因组dna会插入到人基因组当中发生整合。
3.现阶段关于hpv的研究大多集中在hpv e6和e7癌基因的作用,忽略了hpv整合的重要性,大多数病变进展至宫颈癌阶段都出现了hpv基因整合,因此hpv整合与疾病进展过程密切相关,并且是疾病进展的重要标志。hpv整合引起宿主细胞基因组强烈不稳定,致使整合位点附近发生大量的染色体扩增、缺失、重排和易位。与此同时,hpv整合在人类基因组并非随机发生,大部分已知整合位点位于基因组脆性位点和癌基因或者抑癌基因附近,这些整合事件会导致抑癌基因功能异常失活或者癌基因异常激活。
4.整合进人类基因组的hpv病毒可能产生不属于正常细胞的突变蛋白,这些异常蛋白质序列在胞内被蛋白酶体加工成短肽,然后再被人类白细胞抗原结合,呈递到细胞表面上,从而作为外来抗原即新抗原被t细胞识别。新抗原疫苗肿瘤免疫治疗方法以其治疗效果显著、适用癌种广泛和毒副作用小等特点已成为免疫治疗家族重要的成员。因此,基于hpv整合后的人类基因组病毒插入位点开发的新抗原预测算法对宫颈癌等肿瘤的研究和临床应用都具有重要意义。
技术实现要素:
5.针对当前hpv研究中存在的问题,本发明充分考虑人类基因组中的hpv插入片段,开发了一套基于hpv整合的新抗原分析的生物信息学方法。
6.本发明在于公开一种基于hpv整合的肿瘤新抗原预测方法,包括以下步骤:
7.s01,组装肿瘤样品转录本;
8.s02,筛选样本hpv整合转录本;
9.s03,翻译hpv整合转录本为多肽;
10.s04,获取多肽短序列片段,并过滤人类正常蛋白质组中多肽;
11.s05,样品人类白细胞抗原基因分型;
12.s06,肽段亲和力预测、新抗原筛选。
13.优选的,所述样本为新鲜肿瘤组织样品;作为替代,可以选用外周血样品。
14.在本发明的一些实施方式中,s01中,包括以下步骤:
15.s11,建库测序,获取样品mrna的深度测序数据;
16.s12,过滤测序数据,去除测序数据中平均碱基质量低或者包含测序引物接头的短读序列,并将数据格式转换为后续组装软件能够接收的形式;
17.s13,将过滤后的数据组装为转录本。
18.在本发明的一些优选的实施方式中,s11中,采用去核糖体链特异性建库方法和小片段富集筛选建库方法来建库测序。
19.在本发明的一些优选的实施方式中,s11中,样本数据中包括多个重叠或部分重叠的短读序列,获取样品的rna
‑
seq测序数据不小于15g。
20.在本发明的一些优选的实施方式中,s12中,所述平均碱基质量不低于20。
21.在本发明的一些实施方式中,s13中,利用de novo组装软件trinity将过滤后的数据组装为转录本;
22.优选地,使用de novo组装软件trinity时,利用窗口滑动切割reads为kmer,通过相邻kmer之间的连接构建de bruijn图,然后在每幅图中得到所有的剪接异构体代表路径;进一步优选,去除组装过程中的unique occurring kmer。
23.在本发明的一些实施方式中,s02中,先将组装得到的转录本与hpv基因组进行比对,从中筛选出hpv阳性转录本,然后再与人类参考基因组进行比对,从中筛选能同时比对到hpv基因组和人参考基因组的转录本,即hpv整合转录本。
24.在本发明的一些优选的实施方式中,s02中,在分析比对结果时,过滤掉比对长度小于100bp、序列相似度小于98.0%的比对结果。
25.在本发明的一些实施方式中,s03中,包括以下步骤:
26.s31,完整阅读框翻译:若在转录本中找到完整开放阅读框orf,则将找到的orf进行从头翻译;
27.s32,起始密码子翻译:若未能在整合转录本中寻找到orf,则在其中搜寻起始密码子atg,然后从起始密码子开始往后进行翻译,得到多肽片段;
28.s33,外显子翻译:若整合位点在人外显子区域,则利用人类外显子进行辅助翻译;
29.s34,将s31或s32和s33得到的结果文件合并。
30.在本发明的一些优选的实施方式中,s04中,将得到的蛋白/多肽序列分割成长度为8~12的kmer,并过滤人类正常蛋白质组中多肽。
31.在本发明的一些实施方式中,s06中,通过算法预测特定hla亚型与多肽之间的亲和性,筛选出与hla分子亲和性强的肽段;
32.优选地,利用软件netmhc
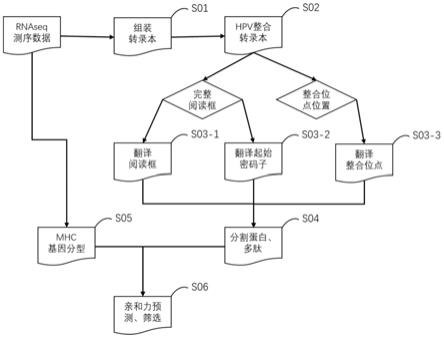
‑
4.0预测分割之后的kmer残基肽段与hla分子的亲和力,选取亲和力大于阈值的为候选新抗原。
33.与现有技术相比,本发明的方案具有如下优势:
34.一、现阶段关于hpv的研究大多集中在hpv基因组本身e6和e7癌基因的作用,忽略了hpv整合的重要性,本发明得到的新抗原是hpv整合的结果,与疾病进展过程密切相关。
35.二、从来源上讲,当前预测新抗原常用的方法是首先识别dna层面的突变,再预测得到肽段。因为dna需要先转录成rna再翻译为蛋白质,由于发生突变的dna片段可能没有发生转录,导致预测得到的新抗原不会产生。本发明得到的新抗原来自于rna测序数据,是细
胞转录的结果,有更高的概率翻译产生蛋白质。
附图说明
36.图1本发明一种实施方式的基于hpv整合的新抗原分析的流程图。
具体实施方式
37.以下通过特定的具体实例说明本发明的实施方式,本领域技术人员可由本说明书所揭露的内容轻易地了解本发明的其他优点与功效。本发明还可以通过另外不同的具体实施方式加以实施或应用,本说明书中的各项细节也可以基于不同观点与应用,在没有背离本发明的精神下进行各种修饰或改变。若非特别之处,实施例和对比例为组分、组分含量、制备步骤、制备参数相同地平行试验。
38.实施例1
39.如图1所示,一种基于hpv整合的新抗原预测方法,该方法包括由处理器执行的如下步骤:
40.s01,组装肿瘤样品转录本
41.具体地,首先采用去核糖体链特异性建库方法和小片段富集筛选建库方法建库测序,获取样品mrna的深度测序数据。样本数据中包括多个重叠或部分重叠的短读序列,重叠程度的不同与测序的深度有关,应获取样品不小于15g的rna
‑
seq测序数据。
42.其次,过滤测序数据,去除测序数据中平均碱基质量低于20或者包含测序引物接头的短读序列,并将数据格式转换为后续组装软件能够接收的形式,具体地对组装软件trinity而言,双端测序的read id分别应为“name/1”和“name/2”。
43.然后利用de novo组装软件trinity将过滤后的数据组装为转录本,软件首先利用窗口滑动切割reads为kmer,通过相邻kmer之间的连接构建de bruijn图,然后在每幅图中得到所有的剪接异构体代表路径。为降低内存消耗并且减少拼接过程中的噪音,可以去除组装过程中的unique occurring kmer。
44.s02,筛选样本hpv整合转录本
45.hpv整合是指hpv基因组dna插入到人基因组dna中,从数据上看,整合后的转录本一部分比对到人基因组,另一部分比对到hpv基因组;由于hpv基因组(约7.9k)相对人基因组(约3g)较小,且相差较大,为减少比对计算资源消耗,先将组装得到的转录本与hpv基因组进行比对,从中筛选出hpv阳性转录本,然后再与人类参考基因组进行比对,从中筛选能同时比对到hpv基因组和人参考基因组的转录本,即hpv整合转录本。
46.参考基因组数据是指各染色体上的碱基序列数据,通常为fasta格式,参考基因组数据可以通过ucsc或者ensemble下载,使用版本为hg38/grch38;一些常见的人基因组文件中可能已经包含hpv病毒基因组序列,如grch38.d1.vd1.fa中就包含有189条hpv相关序列,本实施例中需要选用仅包含primary_assembly的人基因组文件,如
47.homo_sapiens.grch38.dna.primary_assembly.fa。
48.在分析比对结果时,需要过滤掉比对长度较短、相似度较低的比对结果,本实施例要求最低比对长度为100bp,最低序列相似度为98.0%。
49.s03,翻译hpv整合转录本为多肽
50.对hpv整合转录本进行翻译,本实施例分为以下3种情况:
51.s03
‑
1,完整阅读框翻译
52.若在转录本中找到完整开放阅读框(open reading frame,orf),则将找到的orf进行从头翻译;具体地,先利用orffinder在整合转录本中查找开放阅读框,然后则对orf利用密码子表进行翻译。
53.s03
‑
2,起始密码子翻译
54.若未能在整合转录本中寻找到orf,则在其中搜寻起始密码子(atg),然后从起始密码子开始往后进行翻译,得到多肽片段;
55.s03
‑
3,外显子翻译
56.若整合位点在人外显子区域,则利用人类外显子进行辅助翻译。具体来说,本实施例假设hpv序列插入到人基因外显子区域,整合位点前的人外显子序列按原有方式正常翻译,整合位点后的hpv序列紧接着翻译,得到多肽片段。需要注意的是hpv整合转录本的翻译方向由转录本比对人基因组正链还是负链以及比对的蛋白编码基因位于人基因组正链还是负链共同决定,当二者同为正链或者同为负链时,从正向翻译转录本,否者需要先对转录本取反向互补序列,然后再进行翻译。
57.最后,将这三个部分得到的结果文件合并。
58.s04,获取多肽短序列片段,并过滤人类正常蛋白质组中多肽
59.kmer是值一个字符串包含的所有可能的长度为k的子字符串集,对于一条输入蛋白序列而言,从第一个氨基酸残基开始,采用步长为1的滑动窗依次提取固定长度k的序列,这些序列就是kmer。具体地,将上一步得到的蛋白序列分割成长度较小的kmer,利用软件将s03得到的蛋白/多肽序列分割成长度为8~12的kmer,并过滤人类正常蛋白质组中多肽。具体地,uniprot是一个综合性的非冗余数据库,包含所有的公开的人类蛋白质序列,去除在uniprot蛋白质中的kmer片段。
60.s05,样品人类白细胞抗原基因分型
61.人类白细胞抗原(human leukocyte antigen,hla),又被称为人类的mhc(major histocompatibility complex),是控制细胞间相互识别、调节免疫应答的一组紧密连锁基因群。hla位于6号染色体短臂,具有高度的遗传多态性,是基因中等位基因多态性最高的基因复合体。所编码的mhc i类分子主要介导cd8+t细胞对抗原的识别和扑杀,ii类分子则主要与cd4+t细胞结合,从而启动免疫应答。不同亚型hla分子对同一多肽的亲和力可能不同,明确样品hla亚型,可以筛选出亲和力较高的多肽作为候选新抗原。具体的,利用软件seq2hla对样品的人类白细胞抗原进行基因分型。
62.s06,肽段亲和力预测、新抗原筛选
63.肿瘤细胞表达的突变蛋白不被正常细胞表达,这些异常蛋白质序列在胞内被蛋白酶体加工成短肽,然后再被人类白细胞抗原结合,呈递到细胞表面上,从而作为外来抗原被t细胞识别。通过算法预测特定hla亚型与多肽之间的亲和性,筛选出与hla分子亲和性强的肽段。具体地,利用软件netmhc
‑
4.0预测分割之后的kmer残基肽段与hla分子的亲和力,选取亲和力大于阈值的为候选新抗原。
64.得到候选新抗原之后,提取其测序数据覆盖度计算表达丰度,具体地,首先对组装得到的转录本建立索引,再利用比对软件将过滤后的数据回帖到转录本,然后将候选新抗
原片段定位到转录本具体位置,提起特定位置的测序数据支持数,最后根据多肽的相应特征如mhc亲和力、表达丰度等进行新抗原筛选。所用软件具体提参数如下:
65.使用trimmomatic进行原始数据的过滤,其示例命令为:
[0066][0067][0068]
其中sample_1.fastq.gz与sample_2.fastq.gz为输入的原始数据,sample.clean.r1.fq.gz、sample.unpaired.r1.fq.gz、sample.clean.r2.fq.gz和sample.unpaired.r2.fq.gz是输出数据,illuminaclip:adapter.fa:2:30:10:8:true表示切除测序引物序列,参数后面分别接接头序列文件、允许的最大错配数、palindrome模式下匹配碱基数阈值、simple模式下的匹配碱基数阈值;leading指明切除首端碱基质量小于20的碱基;trailing指明切除末端碱基质量小于20的碱基;minlen指明最小的序列长度。
[0069]
使用trinity进行de novo组装,其示例命令为:
[0070][0071]
其中
‑‑
left read1和
‑‑
right read2为过滤后的原始数据,
‑‑
min_kmer_cov 2表示去除组装过程中的unique occurring kmer;
‑‑
output trinity_out_dir为结果文件存放路径,需要注意的是,该路径名必须包含trinity字符。
[0072]
使用blat将组装得到的转录本比对到hpv基因组,从中提取hpv阳性转录本,然后将hpv阳性转录本比对到人基因组,再从中提取hpv整合转录本,其示例命令为:
[0073][0074][0075]
其中assemble.fa为经de novo组装得到的转录本文件,hpv.fa为hpv基因组,homo_sapiens.grch38.dna.primary_assembly.fa为人类参考基因组primary部分序列。
‑
out=blast8表示采用类似blast m8的格式输出结果文件,共包含12列,分别为查询序列id标识、比对上的目标序列id标识、序列比对的一致性百分比、符合比对的比对区域长度、比对区域的错配碱基数、比对区域的碱基间隔数、比对区域在查询序列上的起始位点、比对区域在查询序列上的终止位点、比对区域在目标序列上的起始位点、比对区域在目标序列上的终止位点、比对结果期望值、比对结果的bit score值;
‑
m 100
‑
i 98
‑
g 5表示最低比对长度为100bp,最低序列相似度为98.0%,最大允许间隔为5个碱基。
[0076]
使用软件对hpv整合转录本进行翻译,其示例命令为:
[0077][0078]
其中integration.fa为hpv整合转录本,homo_sapiens.gtf为人类基因组注释文件,用于定位转录本整合位点。
[0079]
使用seq2hla进行mhc基因分型,其示例命令为:
[0080][0081]
使用netmhcpan进行肽段亲和力预测,其示例命令为:
[0082][0083]
其中protein.fa为整合转录本经翻译得到的蛋白质、肽段集合,uniprot.fa为人类蛋白文件。
[0084]
以上对本发明优选的具体实施方式和实施例作了详细说明,但是本发明并不限于上述实施方式和实施例,在本领域技术人员所具备的知识范围内,还可以在不脱离本发明构思的前提下作出各种变化。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1