一种基于全外显子组测序分析HPV病毒整合位点的方法与流程
一种基于全外显子组测序分析hpv病毒整合位点的方法
技术领域
1.本发明属于分子生物学领域,涉及医学、生物技术和生物信息学,具体的是涉及一种高通量测序检测hpv整合位点的方法。
背景技术:
2.宫颈癌是全球女性最常见的妇科恶性肿瘤之一,据统计世界上每年宫颈癌新发患者约53万例,每年死亡约27万人。目前85%的宫颈癌病例发生在发展中国家,其中,中国每年新增发病病例超过13.7万,占全世界总数的28.8%。
3.研究表明,宫颈癌的形成是一个复杂且连续的发展过程,其发展历程可概括为:宫颈上皮内瘤样变(cin-i)
→
cinii
→
cin iii
→
宫颈原位癌(cis)
→
早期宫颈浸润癌
→
宫颈浸润癌(icc)。可见,宫颈上皮癌前病变是宫颈癌的发病起始,而高危型人乳头瘤病毒hpv的持续性感染被证实是导致正常宫颈上皮细胞发生癌病的最主要因素。虽然hpv感染宿主后,既可以游离形式存在,也可整合到宿主基因组中,但大量研究表明宫颈癌的发生和发展总是伴随着hpv整合入宿主基因组的现象。因此,检测hpv整合宿主基因组可作为诊断宫颈癌的有效手段。
4.目前,关于hpv病毒整合入宿主基因组中的研究方法,最常见的是以样本mrna为研究对象,基于pcr扩增技术拓展的各类方法,但rna样本的保存与运输存在一定的难度,同时并不适用大样本的分析和研究。而高通量测序技术以能一次并行对几十万到几百万条dna分子进行序列测定,大大提高了dna的测序效率。其中,相同芯片通量下,全外显子组测序相较于全基因组来说,测序样本数更多,覆盖深度更高,成本更低,耗时更短。
技术实现要素:
5.本发明的目的在于提供一种基于全外显子组测序来分析宿主hpv病毒整合位点的方法,来解决大样本hpv整合位点分析困难、费用昂贵、费时等问题。
6.本发明的方法,包括数据过滤、序列比对、确定整合位点等。
7.本发明基于高通量全基因组测序技术,根据匹配质量及paired_end reads序列间的pe关系评估hpv病毒整合位点的准确性,以多层次水平的测序信息整合分析,最终实现病毒插入位点的精确定位,并进一步完善病毒整合分析的方法
8.宫颈癌组织样本和对应血液样本。
9.测序服务器为华大mgiseq-2000rs fast测序仪。
10.测序使用参考基因组为hgi9。
11.全外显测序panel为安捷伦v7-s31285117_hgs_19。
12.reads测序质量报告分析软件fastqc(v0.11.8)。
13.质量过滤软件fastp(v0.21.0)。
14.构建参考基因组hg19与hpv的索引软件samtools。
15.序列比对参考基因组hg19与hpv基因组。
16.序列比对软件bwa(v0.7.17)。
17.序列比对算法bwa-mem。
18.reads文件的压缩转换及排序软件samtools。
19.使用picard软件(v1.119)删除reads中由pcr扩增产生的非样本基因组中的reads。
20.提取比对到hpv基因组上的reads,使用工具samtools view。
21.通过blastn软件进行一步验证。
22.使用excel表格统计hpv病毒整合位点。
附图说明
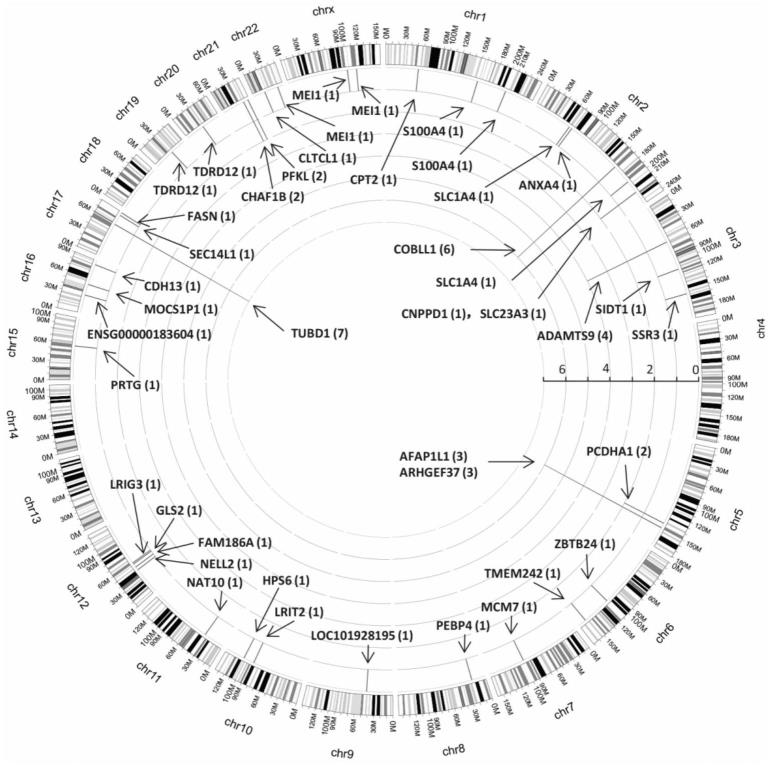
23.图1显示了利用excel表格统计各样本在不同hpv基因组上的插入整合情况,并定位至hpv基因组上的基因,整合位点结果标记在人类基因组上。
具体实施例
24.下面结合具体实施方式进一步阐述本发明的技术方案。
25.样本准备:从不同阶段感染hpv病毒的患者体内取宫颈组织及对应血液样本,对宫颈组织制作石蜡切片与,共52对。
26.样本基因组dna提取:使用商用试剂盒提取宫颈组织及血液样本中的dna。文库构建:磁珠法提取试剂盒(mgieasy,1000006988)进行dna提取及纯化,qubit3.0荧光定量仪(thermofisher,q33216)进行核酸定量。若基因组dna量足够,推荐使用200ng及以上基因组dna(推荐浓度≥15ng/μl)进行文库构建,使用mgieasy酶切dna文库制备试剂盒(mgieasy,v2.0)按照其说明书的表述进行构建文库。bioanalyze(agilent technologies,g2939aa)检测dna片段大小,文库片段主峰应该在430bp附近。qubit3.0荧光定量仪(thermofisher,q33216)进行文库核酸定量,依据文库浓度。将每12个文库等质量混合,形成一个混合文库,取混合文库1500-2000ng,使用agilent sureselect human all exon v7试剂盒捕获全外显子组区域片段,利用使用mgieasy酶切dna文库制备试剂盒(mgieasy,v2.0)进行捕获文库的扩增(post-pcr)、纯化后获得全外显子组文库。
27.高通量测序:将上述构建好的全外显子组文库按照每个lane(fcl芯片共有4个iane)至少投入280ng文库的标准加入到fcl测序芯片中,将测序芯片放入华大mgiseq-2000rs测序仪中,设置pe150的双端测序程序,利用测序服务器为华大mgiseq-2000rs fast测序仪进行测序。获得全外显子测序数据。
28.通过fastqc查看测序数据的质量信息,获取质量报告,判断样本测序是否合格,若不合格,则剔除该样本,避免对后续结果产生影响。
29.根据质量报告使用fastp软件(v0.20.0),删除质量q20<90%的读数,通过强加读取映射的最低phred质量得分(mapq)来执行过滤,删除低映射质量(mapq<5)的读数,去除接头及低于30bp的reads,并去除reads的5
′
端10bp的碱基(gc含量波动较大),完成数据的过滤。
30.在university of california santa cruz(ucsc)中下载人类基因组hg19的fasta序列,以及在ncbi中下载已明确报道的18种高危型hpv(6,11,16,18,31,33,35,39,
45,52,56,58,59,66,68,69,82和83)病毒基因组构成混合病毒库hr-hpv。合并hg19和hr-hpv成一个fasta文件,使用samtools index构建人类hg19和hpv基因组索引。
31.将过滤后高质量的reads使用bwa软件中的bwtsw方法构建序列比对索引:bwa index-a bwtsw。
32.使用bwa软件中mem算法将质控后的reads比对至参考基因组上。获得比对后的sam文件,使用samtools view转化为bam文件,并使用samtools sort对bam文件中reads序列按照染色体进行排序。
33.使用picard软件中的markduplicates工具删除非人类基因组中的reads序列。
34.使用samtools与awk提取出比对至hpv基因组上的所有reads。一类reads可以完全比对上hpv病毒基因组;另一类reads则是一部分比对上hpv病毒基因组,剩余部分比对上人类基因组hg19;最后一类则是一条reads比对至人类基因组hg19,另一条reads比对至hpv病毒基因组上。基于后面这两类hpv病毒宿主嵌合型的reads序列,便能够检测出hpv病毒整合入宿主基因组的准确位置。
35.为验证bwa比对的质量以及确认病毒宿主准确的嵌合序列,所有的嵌合型reads均通过blastn软件进一步比对验证。成对reads的前后序列的比对结果分别输出在两个单独的文件,同样包括比对部分的比对情况、名称、比对质量、位置等信息组合。
36.设定判断整合位点标准:第一,若两条测序reads比对至宿主基因组的位置坐标相差350,且比对病毒基因组位置同样相差350bp以内,则判定为同一整合位点;第二,将在基因组坐标上相差5nt之内的reads兼并为一条序列,计数时则只计一次。
37.利用excel表格统计各样本在不同h pv基因组上的插入整合情况,并定位至hpv基因组上的基因,整合位点结果标记在人类基因组上,见附图1。
38.以上所述,仅为本发明较佳的具体实施方式,本发明实施范围不限于此,任何熟悉本技术领域的技术人员在本发明披露的技术范围内,都可以轻易得到技术方案的简单变化,或等效替换,这些变化或替换均落入本发明的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1