基于个体化代谢模型的肿瘤分型与潜在靶标预测方法与流程
1.本发明涉及癌症亚型分型领域,特别涉及一种基于个体化代谢模型的肿瘤分型与潜在靶标预测方法。其是基于个体化代谢网络模型预测的代谢流变化特征来区分不同的肿瘤病人,进行精准分子分型,并通过计算模拟发现对肿瘤细胞生长具有重要影响的基因作为候选靶标。
背景技术:
2.肿瘤精准医疗的关键在于个性化,针对个人的诊疗措施需要准确的分子分型和准确的预后标志物及治疗靶标。由于恶性肿瘤具有异质性高、预后差、死亡率高、不易早期诊断的特点,目前仍缺少有效的分型和靶向诊疗手段。
3.随着人们对肿瘤生物学及肿瘤代谢复杂性的了解深入,发现癌细胞与正常组织相比存在着大量的代谢异质性,除了瓦博格效应,癌细胞在代谢方面还有很多与正常细胞不同之处。例如p53缺陷结肠癌细胞激活了甲羟戊酸途径来促进泛醌的合成,这对于在代谢受损的环境中维持线粒体电子运输以进行呼吸和嘧啶合成至关重要。研究表明,人类肝脏中线粒体代谢异常是致癌因素之一,肝癌中的免疫检查点调节肿瘤的线粒体代谢能量,肿瘤微环境以及肿瘤特异性免疫反应,该调节在癌细胞和免疫细胞的代谢重编程之间形成正循环。癌症进展与代谢重编程息息相关,在癌症从癌前病变组织发展成局部浸润至转移的过程中代谢表型及代谢依赖性都在发生改变。“代谢重编程”的肿瘤细胞变化现象被认为是癌症的十大标志之一。
4.已有一些基于组学信息的癌症分子分型的方法,如研究者利用早期肝细胞癌蛋白质组学以及磷酸化蛋白质组学的信息将早期肝细胞癌分成三种亚型:s
‑
i、s
‑
ii和s
‑
iii,其中s
‑
iii亚型的最大特点是胆固醇稳态代谢受损。另外,基于蛋白质组数据对hbv诱导的肝癌病人进行分子分型,发现三种亚型具有显著不同的预后生存,并且其蛋白质表达分别富集在代谢重编程、免疫微环境以及细胞增殖的过程。但是,目前还没有基于代谢层次特征对癌症进行分型的方法,特别是对于各种癌症普适性的分析方法。
5.目前癌症的临床分期方案主要有bclc分期、tnm分期等,虽然有客观的衡量标准,但同一分期内患者的异质性依然显著,不同分期的患者不能展现出显著差异的生存状况,影响预后评估和治疗方案的选择。因此,本发明在分子层面对癌症病人进行更加有效的分型,并据此对不同分型的病人实施个体化治疗手段。另外,临床上癌症患者对于靶向药物的有效响应有限,例如肝癌对索拉菲尼、乐菲替尼等一线药物的有效响应仅有30%左右。因此,发现更有效的个体化治疗靶点是肝癌精准医学领域有待突破的瓶颈。
6.另外,目前中国专利公开了一些肿瘤病人的分型方法,例如中国发明专利公开号cn108765411a公开的一种基于影像组学的肿瘤分型方法;中国发明专利公开号cn110400601a公开的基于rna靶向测序和机器学习的癌症亚型分型方法及装置;中国发明专利公开号cn111311558a公开的一种用于胰腺癌预测的影像组学模型的构建方法;中国发明专利公开号cn110415206a公开的一种识别肺腺癌浸润分型的方法;中国发明专利公开号
cn107849613a公开的用于肺癌分型的方法。但是均不是基于代谢层次特征对癌症进行分型的方法。
技术实现要素:
7.本发明所要解决的技术问题在于针对目前肿瘤病人的分型和潜在靶标预测方法所存在的不足而提供一种基于个体化代谢模型的肿瘤分型与潜在靶标预测的方法。其解决了基于代谢层次对癌症进行精准分子分型的挑战,并基于模型的定量模拟方便快捷地预测潜在靶标,为肿瘤精准诊疗提供重要参考。
8.为了实现本发明的目的,本发明所采用的技术方案是:
9.一种基于个体化代谢模型的肿瘤分型与潜在靶标预测方法,包括如下步骤:
10.步骤一:构建肿瘤病人个体化代谢网络模型;
11.步骤二:基于步骤一所述的肿瘤病人个体化代谢网络模型的代谢特征进行精准分子分型;
12.步骤三:基于步骤一所述的肿瘤病人个体化代谢网络模型预测对肿瘤细胞生长具有重要影响的基因作为潜在靶标;
13.所述步骤二和步骤三可以单独运行或者同时运行。
14.在本发明的一个优选实施例中,所述步骤一中构建肿瘤病人个体化代谢网络模型的具体方法是:选用人类全基因组规模代谢网络模型human
‑
gem作为通用模型,将病人的转录组表达数据或蛋白质组表达数据按表达量进行排序,分为高、中、低、不表达的表达谱数据,采用tinit算法将表达谱数据与human
‑
gem模型整合,依据表达情况设置代谢反应流量的约束,从而获得肿瘤病人个体化代谢网络模型。
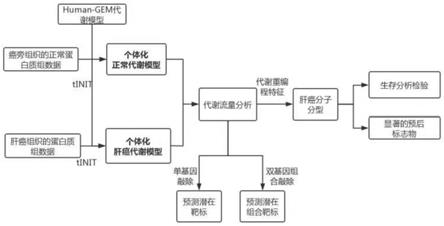
15.在本发明的一个优选实施例中,所述病人的转录组表达数据分为肿瘤组织对应的肿瘤转录组和癌旁组织对应的正常转录组;所述蛋白质组表达数据分为肿瘤组织对应的肿瘤蛋白质组和癌旁组织对应的正常蛋白质组;所述肿瘤病人个体化代谢网络模型分为个体化正常代谢模型和个体化肿瘤代谢模型。
16.在本发明的一个优选实施例中,所述步骤二中的基于步骤一所述的肿瘤病人个体化代谢网络模型的代谢特征进行精准分子分型的具体方法如下:首先,基于模拟得到的代谢流量信息,对肿瘤细胞和正常细胞进行层级聚类,识别能够区分肿瘤和正常细胞的关键代谢特征。然后再对所有肿瘤模型进行层级聚类或k
‑
means聚类,并利用临床数据进行kaplan
‑
meie生存分析的检验,得到具有显著生存差异的分子分型,发现不同亚型之间显著差异的代谢反应和代谢物。
17.在本发明的一个优选实施例中,所述步骤三中的基于步骤一所述的肿瘤病人个体化代谢网络模型预测潜在靶标的具体方法是:对于构建的病人个体化肿瘤组织的代谢网络模型,以肿瘤细胞生长(cell growth)为最优目标函数,进行流量平衡分析得到原始的生长速率grwt,如公式(1):
[0018][0019]
a
j
≤v≤b
j
[0020]
s代表化学计量矩阵,v代表所有反应的代谢流量,即反应速率,s
·
v满足质量守
恒,a
j
和b
j
分别代表反应流量的上限和下限约束;
[0021]
然后将所有的基因逐一做单敲除,得到敲除后的生长速率grko,按公式(2)计算肿瘤细胞的生长速率变化:
[0022]
grratio=grwt/grko
ꢀꢀꢀ
(2)
[0023]
这里grratio的范围是[0,1],通常当比值grratio<=0.5时,认为该基因对于肿瘤细胞为致死基因,能够显著抑制肿瘤细胞的生长。同时,为避免对正常细胞造成损害,要求候选基因不能是正常细胞代谢模型中生长的必需基因,且要求候选基因至少在1/3以上的样本中出现,则可作为潜在的治疗靶标;对于基因组合敲除的模拟与单基因敲除类似,即遍历所有的两两基因组合敲除,得到肿瘤细胞生长的变化比值。
[0024]
在本发明的一个优选实施例中,所述生存分析是利用临床数据进行生存分析的检验,得到具有显著生存差异的分子分型,发现不同亚型之间显著差异的代谢反应和代谢物。
[0025]
在本发明的一个优选实施例中,所述肿瘤为肝癌。
[0026]
由于采用了如上的技术方案,本发明具有如下优点:
[0027]
1.本发明建立的分子分型方案对应的生存曲线差异更为显著。
[0028]
2.基于代谢层次发现的预后标志物能够通过体液或血液进行检测,更加方便、易推广。
[0029]
3.本发明预测的潜在靶标是基于肿瘤细胞生长的定量模拟得到的,不同分型的病人具有个性化靶标,能够提高药物有效性响应。
附图说明
[0030]
图1为基于个体化代谢模型的肿瘤分型与潜在靶标预测方法的流程示意图。
[0031]
图2为肝细胞癌样本的分子分型及生存分析结果示意图。
具体实施方式
[0032]
以下结合附图和具体实施方式来进一步描述本发明。
[0033]
参见图1和图2,本发明的基于个体化代谢模型的肿瘤分型与潜在靶标预测方法,包括如下步骤:
[0034]
步骤一:构建肿瘤病人个体化代谢网络模型;
[0035]
步骤二:基于步骤一所述的肿瘤病人个体化代谢网络模型的代谢特征进行精准分子分型;
[0036]
步骤三:基于步骤一所述的肿瘤病人个体化代谢网络模型预测对肿瘤细胞生长具有重要影响的基因作为潜在靶标。
[0037]
1.构建肿瘤病人个体化代谢网络模型
[0038]
用于个体化代谢网络构建的原始代谢网络模型需要尽量涵盖所有已知的生化反应及通路。选用最新的人类全基因组规模代谢网络模型human
‑
gem模型,与来自tcga等数据库或某项研究检测获得的癌症转录组或蛋白质组表达数据整合,采用tinit算法来构建个体化模型。
[0039]
首先,将病人的转录组表达数据或蛋白质组表达数据按表达量进行排序,转化为高、中、低、不表达四种情况的分类型数据,然后采用tinit算法将表达谱数据与human
‑
gem
模型整合,算法的核心是根据基因或蛋白质的表达情况删除模型中不活跃的基因和反应,同时设置生成的个体化模型需满足的代谢功能任务,如果被删除的基因和反应会引起代谢功能任务执行失败,则这些基因和反应必须要在个体化模型中保留。针对每一个病人的癌与癌旁组织的表达谱数据,按以上方法分别构建特异性癌细胞与癌旁细胞的代谢模型。
[0040]
2.基于个体化代谢模型的代谢变化特征进行分子分型
[0041]
首先,基于模拟得到的代谢流量信息,对肿瘤细胞和正常细胞进行层级聚类,识别能够区分肿瘤和正常细胞的关键代谢特征。然后再对所有肿瘤模型进行层级聚类或k
‑
means聚类,并利用临床数据进行kaplan
‑
meie生存分析的检验,得到具有显著生存差异的分子分型,发现不同亚型之间显著差异的代谢反应和代谢物。图2展示的是以肝细胞癌为例的分型及生存分析的结果。
[0042]
然后,进一步从exchange metabolite中筛选潜在的代谢标志物,这些代谢物可以在胞外进行检测,相对而言临床上更容易被检测到。通过正交偏最小二乘法opls分析肿瘤样本与癌旁样本之间存在差异的exchange反应,确定这部分反应交换的代谢物,选择变量投影重要度vip>1或2(具体肿瘤依数据而定)的交换代谢物作为区分癌症与癌旁的标志物。再对于不同分型的癌症样本进行opls分析及vip值筛选,获得显著区分不同生存病人的预后标志物。
[0043]
3.基于个体化代谢模型预测对肿瘤细胞生长具有重要影响的基因作为潜在靶标
[0044]
对于构建的病人个体化肿瘤组织的代谢网络模型,以肿瘤细胞生长(cell growth)为最优目标函数,进行流量平衡分析得到原始的生长速率grwt,如公式(1)。
[0045][0046]
a
j
≤v≤b
j
[0047]
s代表化学计量矩阵,v代表所有反应的代谢流量,即反应速率,s
·
v满足质量守恒,a
j
和b
j
分别代表反应流量的上限和下限约束。
[0048]
然后将所有的基因逐一做单敲除,得到敲除后的生长速率grko,按公式(2)计算肿瘤细胞的生长速率变化:
[0049]
grratio=grwt/grko
ꢀꢀꢀ
(2)
[0050]
这里grratio的范围是[0,1],通常当比值grratio<=0.5时,认为该基因对于肿瘤细胞为致死基因,能够显著抑制肿瘤细胞的生长。同时,为避免对正常细胞造成损害,要求候选基因不能是正常细胞代谢模型中生长的必需基因,且要求候选基因至少在1/3以上的样本中出现,则可作为潜在的治疗靶标。对于基因组合敲除的模拟与单基因敲除类似,即遍历所有的两两基因组合敲除,得到肿瘤细胞生长的变化比值。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1