一种用于基因组碱基变异检测的数据分析系统及分析方法与流程
1.本发明属于生物信息学领域,更具体地涉及一种用于基因组碱基变异检测的数据分析系统及分析方法。
背景技术:
2.第二代测序技术的发展为生命科学研究领域带来了革命性的突破,使得研究人员能够快速方便地获取基因组序列数据,从而为理解生命机制、实现精准医疗提供了前所未有的机遇。然而,如何快速分析这些海量的测序数据,以便服务于临床诊断与治疗,成为生物研究人员急需解决的难题。
3.目前,在一般配置的集群服务器上,计算一个测序深度30x的人全基因组数据,通常需要30~40个小时,远跟不上测序数据产生的速度,因此需要提高全基因组数据分析效率。
技术实现要素:
4.为了解决上述技术问题,本发明提出了一种用于基因组碱基变异检测的数据分析方法,包括如下步骤:
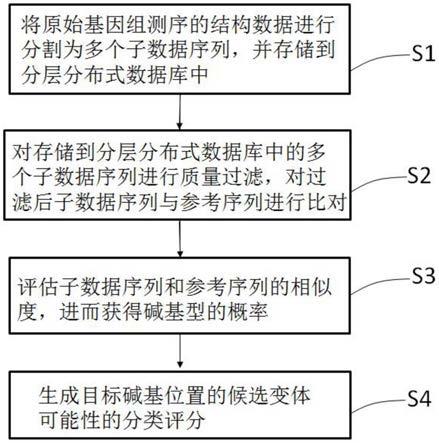
5.s1,将原始基因组测序的结构数据进行分割为多个子数据序列,并存储到分层分布式数据库中;
6.s2,对存储到分层分布式数据库中的多个子数据序列进行质量过滤,并对过滤后的子数据序列与参考序列进行比对;
7.s3,评估子数据序列和参考序列的相似度,进而获得碱基型的概率;
8.s4,生成目标碱基位置的候选变体可能性的分类评分。
9.进一步地,所述步骤s1中,采用特征提取分割方法分割结构数据,按照设定的长度进行分割,使用分割后序列的reads的id作为索引,将链标号、碱基序列、质量作为值,将拥有相同索引的序列组装成一个子数据序列,计算每个子数据序列的长度,基于所述子数据序列的长度,将不同索引的子数据序列存储到分布式数据库中的多个存储服务器中。
10.进一步地,采用以下smith
‑
waterman公式计算分值:
11.m=r*c;
12.r=a*l2+b;
13.其中,m表示smith
‑
waterman计算分值,r为参考序列的长度,c表示对各子数据序列的长度,l表示子存储服务器存储的数据的长度,a和b表示常数。
14.进一步地,所述步骤s3具体包括如下步骤:
15.s31,定义活跃区,根据测序数据的序列比对的数据,识别至少一个活跃区;
16.s32,通过应用de
‑
bruijn图和哈希表,将参考序列作为模板,为每个活动区域构建组装图,依次读取各段reads与图表的一段进行匹配,通过动态生成节点的方式生成所有可能形成的路径,并筛选出有良好支持的路径来表示可能的单倍型;
17.s33,将生成的基础数据加入概率计算队列;
18.s34,使用pair hmm算法对序列进行重新比对,并结合计算分值m得到每个单倍型的概率;
19.s35,采用贝叶斯定理计算碱基型的概率。
20.进一步地,基于贝叶斯全概率公式,在观察到数据d的情况下,基因型g的概率可以这样得到:
[0021][0022]
其中,对于常见的二倍体来说,i等于1,2,3,即有
[0023]
g
i
=h1h2∈{aa,aa,aa};
[0024]
p(g)代表该碱基型的概率,它是我们通过结合参考基因的错误率,snp为纯合子的概率,snp为杂合子的概率以及转换与颠换的概率之比的研究之后,推算得到的概率。
[0025]
p(d/g)是给定基因型g的条件下,出现观测数据d的概率,其值由步骤3求出的单倍体似然值p(d
j
/h
n
)带入以下公式得出:
[0026][0027]
进一步地,针对新增的序列,根据链标号、碱基序列或质量作为值判断其对应的索引,从而加入其所对应的子数据序列,并存储到分布式数据库中的存储服务器中。
[0028]
本发明还提出了一种用于实现数据分析方法的数据分析系统,包括:
[0029]
结构数据获取单元,用于获取原始fastq文件中的基因组测序的结构数据;
[0030]
分割和存储单元,其电连所述结构数据获取单元,用于将所述结构数据分割为多个子数据序列,并存储到分层分布式数据库中;
[0031]
序列比对单元,其电连所述分层分布式数据库,用于对存储到分层分布式数据库中的多个子数据序列进行质量过滤,并对过滤后的子数据序列与参考序列进行比对;
[0032]
概率评估单元,其电连所述序列比对单元,用于评估子数据序列和参考序列的相似度,进而获得基因分型的概率;
[0033]
分类评分单元,其电连所述概率评估单元,用于产生目标碱基位置的变体可能性分类评分。
[0034]
进一步地,所述序列比对单元还包括校准子单元,用于评估子数据序列比对质量以及校准其正确性。
[0035]
进一步地,分类评分单元利用深度卷积神经网络模型输出变体可能性的分类评分,所述深度卷积神经网络模型包括输入序列的表征层,提取特征的上卷积层和下卷积层,特征整合的全连接层以及最后连接的分类器。
[0036]
名词解释:
[0037]
基因是指携带有遗传信息的dna或rna序列(即基因是具有遗传效应的dna或rna片段),也称为遗传因子,是控制性状的基本遗传单位。基因通过指导蛋白质的合成来表达自己所携带的遗传信息,从而控制生物个体的性状表现。
[0038]
基因测序是一种新型基因检测技术,从血液或唾液中分析测定基因全序列,从而预测罹患多种疾病的可能性、个体的行为特征及行为合理等。
[0039]
reads:是一小段短的测序片段,是高通量测序仪产生的测序数据,对整个基因组进行测序,就会产生成百上千万的read,然后将这些read拼接起来就能获得基因组的全序列。
[0040]
序列比对分析:ngs测序下来的短序列(read)存储于fastq文件里面,虽然它们原本都来自于有序的基因组,但在经过dna建库和测序之后,文件中不同read之间的前后顺序关系就已经全部丢失了。因此,fastq文件中紧挨着的两条read之间没有任何位置关系,它们都是随机来自于原本基因组中某个位置的短序列而已。因此,我们需要先把这一大堆的短序列捋顺,一个个去跟该物种的参考基因组比较,找到每一条read在参考基因组上的位置,然后按顺序排列好,这个过程就称为测序数据的比对。
[0041]
比对算法:序列比对的计算方法一般分为两类:全局性比对(global alignments)和局部比对(local alignments)。计算一个全局性的路线,是一个全局优化的形式,其强制按照整个长度的所有查询序列对齐。与此相反,局部比对只确定局部的相似而整个长序列却往往大相径庭。局部比对往往是可取的,但可能更难以计算,因为还有来自确定其他相似区域的挑战。各种计算算法已应用于序列比对问题中,包括缓慢但正规的像动态规划的优化方法、高效率但不彻底的启发式算法,或大型数据库搜索设计的概率方法。
[0042]
smith
‑
waterman:是一种进行局部序列比对(相对于全局比对)的算法,用于找出两个核苷酸序列或蛋白质序列之间的相似区域。该算法的目的不是进行全序列的比对,而是找出两个序列中具有高相似度的片段。
[0043]
哈希表:又称散列算法、哈希函数,是一种从任何一种数据中创建小的数字“指纹”的方法。散列函数把消息或数据压缩成摘要,使得数据量变小,将数据的格式固定下来。该函数将数据打乱混合,重新创建一个叫做散列值(hash values,hash codes,hash sums,或hashes)的指纹。散列值通常用一个短的随机字母和数字组成的字符串来代表。好的散列函数在输入域中很少出现散列冲突。在散列表和数据处理中,不抑制冲突来区别数据,会使得数据库记录更难找到。
附图说明
[0044]
图1为本发明的用于基因组结构变异检测的数据分析方法流程图;
[0045]
图2为本发明的步骤s3的具体实现细节流程图;
[0046]
图3为本发明的数据分析系统的结构示意图;
具体实施方式
[0047]
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0048]
在本技术中,术语“包括”、“包含”或者其任何其他变体意在涵盖非排他性的包含,从而使得包括一系列要素的过程、方法、物品或者设备不仅包括那些要素,而且还包括没有明确列出的其他要素,或者是还包括为这种过程、方法、物品或者设备所固有的要素。在没有更多限制的情况下,由语句“包括一个......”限定的要素,并不排除在包括所述要素的
过程、方法、物品或者设备中还存在另外的相同要素。
[0049]
为解决以上问题,本发明提出了一种用于基因组结构变异检测的数据分析系统及分析方法。
[0050]
参考图1所示,为本发明的用于基因组结构变异检测的数据分析方法流程图,该数据分析方法主要包括四个主要的处理过程:建立分布式存储数据库,将原始基因组测序的结构数据进行分割和存储;测序数据的序列比对;基因分型的概率计算以及分类评分。
[0051]
首先,在建立分布式存储数据库,将原始基因组测序的结构数据进行分割和存储的过程中,具体地包括:
[0052]
获取原始fastq文件中的基因组测序的结构数据;
[0053]
基于特征提取分割原则,将所述结构数据分割为多个子数据序列;
[0054]
需要注意的是,对于基因组结构数据的分割和计算必须在生物学意义下确保计算准确性,同时调节系统负载均衡。比如在变异检测前,数据切分按照染色体区域等生物学依据进行有效切割,调节计算负载的均衡程度,并合理设置重叠距离,以确保数据切割不会导致信息丢失。通过在高通量计算环境中进行合理的数据切割,以此来提高数据传输效率。
[0055]
具体地,使用mapreduce框架对结构数据进行相应的分割,在分割结构数据时采用特征提取(fx)分割方法,该方法通过重写recordreader抽象类将测序序列的双链数据按照hadoop设定的长度进行分割,并将其发送到不同的map进行处理,在每个map中使用分割后序列的reads的id作为索引key,并将链标号、碱基序列、质量作为值value,分割完成后利用reduce的sort功能将拥有相同的索引key的序列组装成一个子数据序列,计算每个子数据序列的长度,基于所述子数据序列的长度,将不同索引key的子数据序列存储到分布式数据库中的多个存储服务器中。
[0056]
由于每个子数据序列的长度存在一定差异,在数据存储时,根据尽可能平均分配的原则,将不同子数据序列分配存储至不同的存储服务器,以保证负载均衡。
[0057]
在优选的实施例中,每个存储服务器所分配得到的多个子数据序列的总的长度是相同的。
[0058]
针对新增的序列,根据链标号、碱基序列或质量作为值value判断属于哪个索引key的子数据序列,从而加入其所对应的子数据序列,并存储到分布式数据库中的存储服务器中。
[0059]
在将所有的子数据序列写入分布式数据库之后,即完成分布式数据库的构建和存储过程。
[0060]
在优选实施例中,在以上数据结构的分割和存储方式中,如果结构数据中的某一特定节点具有较多的关联节点,可优先采用最小切割原则,该特定节点与关联节点的关系都不会被切割,因而该特定节点以及相关联的节点连同其关系均被划分到同一存储服务器,由此保证了一个服务器节点中的数据具有相对较强的内部关联性;且具有良好的扩展性和吞吐能力。
[0061]
接下来,需要测序数据的序列比对,具体地包括:
[0062]
将存储在分布式数据库上的分割好的测序结构数据发送至集群各从机进行处理。在优先实施例中,序列比对前需要对测序结构数据进行质量过滤,过滤掉质量较低的reads,以便去除测序结构数据中的干扰数据,然后使用比对工具对过滤后的序列进行比
对。
[0063]
将参考序列按照染色体组进行相应的分割并分别建立索引。在使用bowtie2比对时,将测序结构序列分别与各染色体组的参考序列进行比对,得到该测序结构序列与各染色体参考序列的比对结果,同时将其保存至分布式数据库相应的结果目录下。
[0064]
序列比对过程可以得到一系列的统计数据,包括对齐读取的总数、错配个数、空隙大小等,这些信息可以用来评估数据的质量以及校准其正确性。
[0065]
在序列比对过程中,利用smith
‑
waterman算法对子数据序列进行打分,将各分批打分结果进行整合运算,得到最终比对结果,实现测序结构序列与参考基因组的精细比对;
[0066]
采用以下smith
‑
waterman公式计算分值:
[0067]
m=r*c
[0068]
r=a*l2+b
[0069]
其中,m表示smith
‑
waterman计算的分值,r为参考序列的长度,c表示对各子数据序列的长度,l表示每个存储服务器存储的数据的长度,a和b表示常数。
[0070]
接下来,进行评估测序结构序列和参考序列的相似度,进而获得可能碱基型的概率。
[0071]
具体地,参照图2所示,对实现细节进行详细阐述:
[0072]
步骤s31,定义活跃区,根据测序数据的序列比对的数据,识别至少一个活跃区;
[0073]
在与参考基因组比对过后,通过遍历测序结构数据来鉴别很有可能发生变异的活跃区域,并传输到下一个步骤中。而在预期噪声水平范围内,没有显示出变异的区域则将在后序步骤中直接跳过,以加快分析的速度。
[0074]
步骤s320,通过应用de
‑
bruijn图和哈希表,可以将参考序列作为模板,为每个活跃区构建组装图。然后,依次读取各段reads与图表的一段进行匹配,通过动态生成节点的方式生成所有可能形成的路径,并筛选出有良好支持的路径来表示可能的单倍型。
[0075]
步骤s330,在得到生物体可能的单倍型之后,我们需要对其不同单倍型的可靠性进行一个评估。在单倍型检测中,这一步通常使用pair hmm算法(隐马氏匹配算法)对序列进行重新比对,并结合其数据质量信息得到每个单倍型的概率。
[0076]
步骤s340,采用贝叶斯定理计算碱基型的概率;
[0077]
具体地,基于贝叶斯全概率公式,在观察到数据d的情况下,基因型g的概率可以这样得到:
[0078][0079]
其中,对于常见的二倍体来说,i等于1,2,3,即有
[0080]
g
i
=h1h2∈{aa,aa,aa};
[0081]
p(g)代表该碱基型的概率,它是我们通过结合参考基因的错误率,snp为纯合子的概率,snp为杂合子的概率以及转换与颠换的概率之比的研究之后,推算得到的概率。
[0082]
p(d/g)是给定基因型g的条件下,出现观测数据d的概率,其值由步骤3求出的单倍体似然值p(d
j
/h
n
)带入以下公式得出:
[0083]
[0084]
这样,我们就得到了给定观测数据下,各位点每种可能的碱基型的概率。
[0085]
最后,进行分类评分。
[0086]
每个碱基型的概率是一个代表一个候选变异位点的特定属性的数字。将概率序列值直接提供给深度卷积神经网络模型。
[0087]
如图所示,深度卷积神经网络模型结构包括输入序列的表征层,提取特征的上卷积层和下卷积层,特征整合的全连接层以及最后连接的分类器。
[0088]
将概率序列输入到表征层,提取特征的上卷积层和下卷积层给出多个向量序列;全连接层将上卷积层和下卷积层给出多个向量序列进行组合,即进行编码或构建;
[0089]
分类层根据全连接层编码或构建后的结构产生目标碱基位置的每个候选变体是真变体还是假变体的可能性的分类评分。
[0090]
在其它实施方式中,所述分类层可以产生所述目标碱基位置的每个候选变体是纯合变体、杂合变体、非变体还是复杂变体的可能性的分类评分。
[0091]
深度卷积神经网络模型需要根据一个更大的训练集对所述分类器进行训练,这样,其性能能够得到进一步提高。
[0092]
如图3所示,为本发明实现上述数据分析方法的数据分析系统,包括:
[0093]
结构数据获取单元,用于获取原始fastq文件中的基因组测序的结构数据;
[0094]
分割和存储单元,其电连所述结构数据获取单元,用于将所述结构数据分割为多个子数据序列,并存储到分层分布式数据库中;
[0095]
序列比对单元,其电连所述分布式数据库,用于对存储到分布式数据库中的多个子数据序列进行质量过滤,并对过滤后的子数据序列进行比对;
[0096]
在优选实施例中,序列比对单元还包括校准子单元,用于评估子数据序列比对质量以及校准其正确性;
[0097]
概率评估单元,其电连所述序列比对单元,用于评估子数据序列和参考序列的相似度,进而获得基因分型的概率;
[0098]
分类评分单元,其电连所述概率评估单元,用于产生目标碱基位置的变体可能性分类评分。
[0099]
分类评分单元利用深度卷积神经网络模型输出变体可能性的分类评分,所述深度卷积神经网络模型包括输入序列的表征层,提取特征的上卷积层和下卷积层,特征整合的全连接层以及最后连接的分类器。
[0100]
此外,上述附图仅是根据本发明示例性实施例的方法所包括的处理的示意性说明,而不是限制目的。易于理解,上述附图所示的处理并不表明或限制这些处理的时间顺序。另外,也易于理解,这些处理可以是例如在多个模块中同步或异步执行的。
[0101]
本领域技术人员在考虑说明书及实践这里公开的发明后,将容易想到本发明的其他实施例。本发明旨在涵盖本发明的任何变型、用途或者适应性变化,这些变型、用途或者适应性变化遵循本发明的一般性原理并包括本发明未公开的本技术领域中的公知常识或惯用技术手段。说明书和实施例仅被视为示例性的,本发明的真正范围和精神由权利要求指出。
[0102]
应当理解的是,本发明并不局限于上面已经描述并在附图中示出的精确结构,并且可以在不脱离其范围进行各种修改和改变。本发明的范围仅由所附的权利要求来限。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1