一种融合DNA形状特征的转录因子结合位点预测方法
一种融合dna形状特征的转录因子结合位点预测方法
技术领域
1.本发明属于生物信息学领域,结合了结构生物学和基因组学的知识设计并实现了一套融合dna形状特征的转录因子结合位点预测新方法。
背景技术:
2.转录因子(tf)可以通过与调节转录的基因组区域结合来协调许多基因的表达。细胞机制利用这些主调节器来调节关键的细胞过程并适应环境刺激。事实上,tf的序列或数量的改变可能是遗传性疾病,复杂疾病,自身免疫缺陷和癌症的主要原因。tf如何与特定的dna调节序列(称为tf结合位点,或简称tfbs,如启动子,增强子)结合以协同调节基因转录和蛋白质合成是一个十分重要的过程,它在许多生物过程中起着关键作。过去十年中,已经产生了大量的免疫沉淀及其高通量测序(chip
‑
seq)数据,并用于研究这些调控过程背后的机制,但由于该方法是tf特异性的,即特定于某一种tf来确定其dna序列上的结合位点序列,以及其高实验成本等原因,不可能在所有细胞类型中分析每个tf结合图谱,因此,需要一个精确的计算方法来解码底层绑定规则。当然,如何预测dna序列中的tfbs是生物信息学中的一个基本问题。
3.转录因子的dna结合特异性是基因调控过程的关键组成部分,但对于tf与其基因组靶位点的高度特异性结合的基础机制知之甚少。早期研究中,我们假设dna转录因子的结合位点完全由碱基序列确定。基于位置权重矩阵(pwm)的方法在dna
‑
蛋白质结合过程建模中取得了巨大成功。后来,gkm
‑
svm(即缺口k
‑
mers和支持向量机)显示出优于基于pwm的方法。近几年,卷积神经网络,加上dna序列的单热编码格式,引起了对预测tfbs的极大兴趣。然而,仅使用初级dna序列预测或插入tfbs已被证明不足以充分建模其底层绑定规则。显然,如果要真正提高预测准确性,就需要改进其底层的建模方式,这一过程是后续预测工作的重要保障。
4.事实上,过去十年的技术发展促进了对许多tf的dna结合偏好的表征的发现与研究。最近的高通量研究强调,tf
‑
dna结合不仅仅依赖于核苷酸序列偏好,并且已经确定了多种相关因素。越来越多的证据支持序列背景,包括侧翼序列和dna形状,在调节序列识别中的广泛贡献。相互作用的辅因子和tf也可以改变序列偏好。除此之外,一些特定于细胞类型的信息,这里主要包括染色质可及性和组蛋白修饰也对tfs与其靶位点的结合有很大影响。
5.在此背景下,越来越多的研究倾向于采用将dna基序与其他特征(包括组蛋白修饰、染色质可及性以及细胞类型等)相结合的方式进行建模。且对不同方法进行过尝试。例如有方法使用不受控制的方法,如分层混合模型或隐马尔可夫模型,以使用染色质可及性数据识别转录因子足迹。他们使用序列基序分数来将足迹归因于不同的转录因子。更近期的方法使用矩阵完备(matrix completion)的方法来完成转录因子结合预测,即使用表示基因组位置,细胞类型和tf结合的3模式张量来推断tf结合。该方法不依赖于序列特异性,但是只能在具有许多chip
‑
seq数据集的充分研究的细胞类型中预测tf结合。值得关注的是,卷积神经网络模型中将序列与表观基因组数据相结合用来预测转录因子结合位点。其
预测过程除了dna序列外,还使用了组蛋白修饰和染色质可及性信息。虽然与只考虑基序信息的同类模型相比有所改进,但是这种方法仅使用了15种细胞类型的标准化dnase
‑
seq数据和5种特定核心组蛋白修饰的信息进行训练和验证,这可能会导致训练模型仅对该特定细胞环境下的tf结合偏好预测良好。综上,目前甚少有人尝试将dna的三维结构特征结合到tfbs的预测建模中。
技术实现要素:
6.本发明要解决的技术问题在于提供一种融合dna形状特征的转录因子结合位点预测方法,所述方法首先构建一个同时包含dna序列基序信息以及dna三维形状信息的可用于转录因子集合位点预测的特殊数据集;然后,提出一种新颖的可以同时融合dna形状特征与序列信息的转录因子结合位点预测模型,所述模型可以将dna的结构特征与dna序列信息相结合,从而提高dna转录因子结合位点预测的准确性。
7.本发明是通过如下技术方案来实现的:
8.一种融合dna形状特征的转录因子结合位点预测方法,所述方法的具体步骤如下所示:
9.1)根据现有技术公开的信息,设计并构建一个具有dna形状特征数据和dna序列信息的特殊数据集,针对dna形状特征的获取采用ht
‑
mc方法预测dna的各种重要结构特征,预测的特征包括小沟宽(minor groove width,mgw),滚动(roll),螺旋桨扭曲(propeller twist,prot)和螺旋扭曲(helix twist,helt);
10.2)dna序列基序数据及dna形状特征数据预处理
11.3d dna形状特征使用基于五聚体的模型预测,该模型基于dna结构的全原子蒙特卡洛模拟建立;输入数据分为两部分为序列和形状;对于dna序列部分,输入是4
×
l的矩阵,其中l是序列的长度,序列中的每个碱基对a、c、t、g分别被表示为四个独热编码[1,0,0,0],[0,1,0,0],[0,0,1,0]和[0,0,0,1];对于dna的形状特征部分,输入是4
×
l的矩阵,其中l是序列的长度,dna序列的形状特征(mgw、roll、prot、helt)被分别描述为每个核苷酸位置的一个通道载体;
[0012]
3)基于cnn的融合dna形状特征的转录因子结合位点预测新模型
[0013]
在收集每个样本的dna序列,dna形状特征(dss),标签数据和编码特征后,确定训练数据的模型为序列+dss模型,序列+dss模型同时使用序列和dss两种类型的数据组合成一个综合模型进行预测;所述的序列+dss模型是基于深度学习中卷积神经网络,采用双输入并行卷积架构,输入为两个4
×
l的矩阵,分别为基因的序列信息矩阵和形状信息矩阵,然后分别进行卷积以及全局最大池化,其中卷积核数为128,卷积窗口大小为1*24,最后将针对两类数据的池化结果连接起来,作为全连接层的输入,神经元数量为32或64,同时使用dropout方法,参数设置为0.1,0.5,0.75,最终输出层神经元数为2,输出阶段使用的激活函数为softmax回归;
[0014]
4)使用步骤2)中预处理后的数据对步骤3)中所述的预测新模型进行训练。
[0015]
作为优先的技术方案,模型的训练过程中使用交叉熵作为损失函数,并使用标准误差反向传播算法和adadetla方法训练模型,将batch_size设为100,并在每个epoch之后验证模型,然后使用早停技巧来停止训练。
[0016]
本发明与现有技术相比的有益效果:
[0017]
1、构建了包含dna形状特征及dna序列信息的特殊数据集,在传统转录因子预测的数据集基础之上添加了对应的dna形状信息。
[0018]
该数据集在传统仅包含序列基序与标签信息的基础之上,还增加了对应于原有序列信息的dna形状特征信息。我们针对dna形状特征信息的获取方法,以及处理该类信息以适应cnn模型等方面,进行深入探讨与研究。形成的通用数据集可用于其他结合dna形状信息与序列信息进行转录因子结合位点预测的研究。
[0019]
2、设计并实现了使用cnn结合dna序列与形状数据预测转录因子结合位点的新模型。
[0020]
模型采用了新型cnn融合框架,结果证明其成功学习到dna的形状信息并将其融合到转录因子结合位点预测的任务中。与其他现有融合dna形状特征的深度学习模型相比,本模型设计复杂度低,训练时间短,可用性强,且与传统融合dna形状特征的数学模型相比,本模型预测准确度更高。
附图说明
[0021]
图1为本发明的dna形状特征类型示意图;
[0022]
图2为本发明的使用cnn结合dna序列与形状信息预测tfbs的统一框架;
[0023]
图3本发明基于深度学习中卷积神经网络的双输入并行卷积架构;
[0024]
图4为基于keras的融合dna形状特征的tfbs预测模型框架图;
[0025]
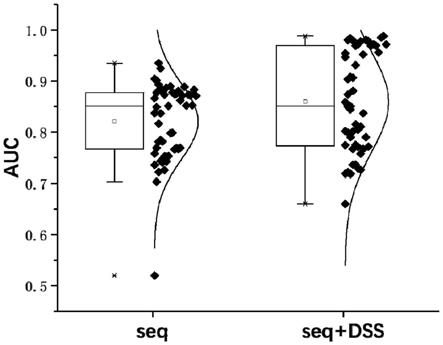
图5基于序列的模型和基于序列与形状信息的模型实验数据分布的比较。
具体实施方式
[0026]
下面通过实施例结合附图来对本发明的技术方案做进一步解释,但本发明的保护范围不受实施例任何形式上限制。
[0027]
实施例1
[0028]
一种融合dna形状特征的转录因子结合位点预测方法,所述方法的具体步骤如下所示:
[0029]
1、数据集的构建
[0030]
首先深入研究蛋白质
‑
dna结合的底层机制的相关科研进展,同时,总结目前dna转录因子结合位点预测的研究进展与现状,搜集调查该领域主流数据集来源信息。其次,针对融合dna形状特征与序列信息的转录因子结合位点预测模型,深入研究dna形状特征获取方法的相关进展,以及dna转录因子结合位点预测相关数据集构造方法,设计并构建具有dna形状特征数据和dna序列信息的特殊数据集。
[0031]
采用ht
‑
mc方法进行dna形状特征的获取,以前的研究通过减少系统中的自由度来提高构象采样的效率。这里的高通量方法旨在预测dna的各种重要结构特征,且基本上可以适应任何长度或数量的序列。该方法可以提高准确性。鉴于它们在dna形状读数中的重要性,预测的特征包括小沟宽(minor groove width,mgw),滚动(roll),螺旋桨扭曲(propeller twist,prot)和螺旋扭曲(helix twist,helt),如图1所示。
[0032]
大量实验和计算数据的广泛验证证明了ht
‑
mc方法的稳健性,在dna形状网络服务
器下面的高通量方法可用于在单个处理器上以不到1分钟的时间完成核苷酸分辨率的整个酵母基因组的dna结构特征预测。
[0033]
本实施例构建了69组转录因子的chip
‑
seq实验数据从encode(http://hgdownload.cse.ucsc.edu/goldenpath/hg19/encodedcc/wgencodeawgtfbsuniform/)下载获得。每组实验数据为fasta格式,且分为训练数据集和测试数据集两部分。在数据集中,给出了dna序列及其对应标注信息。阳性和阴性样本具有相同的gc数量和序列长度(101bp)。然后基于已有方法生成了本实施例中使用的dna形状特征(dss)(包括mgw、roll、prot、helt),该方法基于一个从数千个全原子蒙特卡洛模拟得到的五聚体查询表,且经过x射线和核磁共振结构验证。
[0034]
根据序列数据集中样本的位置,从dss中提取相应位置的信号值。其中可以认为每个核苷酸位置都具有相应的表征每个dna形状特征的值。因此,tfbs和非tfbs被描述为两种类型的特征:(1)用于dna序列信息的独热表征;(2)用于dna形状信息的dss表征。对于每个数据集,使用70%的样本进行训练,10%的样本用于校验,20%的样本用于测试。
[0035]
2、dna序列基序数据及dna形状特征数据预处理。
[0036]
3d dna形状特征使用基于五聚体的模型(ht
‑
mc)预测,该模型基于dna结构的全原子蒙特卡洛模拟建立。四种不同的形状特征,包括小沟宽(mgw),滚动(roll),螺旋桨扭曲(prot),以及螺旋扭曲(helt),且已证明这四类特征在特定情况下对蛋白质
‑
dna结合位点识别具有重要作用。
[0037]
评估的卷积神经网络架构如图3,其中输入分为两部分序列和形状。对于dna序列部分,输入是4
×
l的矩阵。其中l是序列的长度,本实施例中为101bp。序列中的每个碱基对a、c、t、g分别被表示为四个独热编码[1,0,0,0],[0,1,0,0],[0,0,1,0]和[0,0,0,1]。对于dna的形状特征部分,输入是4
×
l的矩阵,其中l是序列的长度。dna序列的形状特征(mgw、roll、prot、helt)被分别描述为每个核苷酸位置的一个通道载体。本实施例中使用101bp的dss数据,则样本的载体大小为1
×
101,又由于本实施例使用了四类dna形状特征,因此其大小为4
×
101。dss是描述dna表观3d特征的连续属性,可能与特定tf的结合有关。本实施例中使用的dna形状特征是单碱基分辨率的数据。
[0038]
从数据角度来看,为了在统一的深度学习框架中结合dss和序列特征,在收集每个样本的dna序列,dss数据,标签数据和编码特征后,本实施例首先实施了两种不同的模型:(1)序列cnn模型,使用dna序列作为特征;(2)dss_cnn模型,使用dss数据作为特征。本实施例中cnn由输入层,卷积层,最大池化层,完全连接层,dropout层以及输出层组成。对于cnn模型,本实施例中内核的数量为128,内核窗口的大小为1*24,完全连接层中的神经元数量设置为64以使模型达到最佳效果。如图3所示,模型基于深度学习中卷积神经网络(convolutional neural networks,cnn),采用双输入并行卷积架构,图4中展示基于keras的融合dna形状特征的tfbs预测模型框架图,首先输入为两个4
×
101的矩阵,分别为基因的序列信息矩阵和形状信息矩阵。然后分别进行卷积(卷积核数为128,卷积窗口大小为1*24),以及全局最大池化,最后将针对两类数据的池化结果连接起来,作为全连接层的输入(这里神经元数量为64),同时使用dropout方法,参数设置为(0.1,0.5,0.75),最终输出层神经元数为2,输出阶段我们使用的激活函数为softmax回归。
[0039]
在设置每种类型数据(这里指dna序列数据与dna形状数据两类)的适当模型,超参
数后,本实施例对比研究了两种不同模型的性能:(1)序列模型,仅使用dna序列数据作为特征;(2)序列+dss模型,同时使用序列和dss两种类型的数据组合成一个综合模型作为特征。
[0040]
对于训练过程,本实施例使用交叉熵作为损失函数。鉴于所选用的损失函数和不同的超参数,使用了标准误差反向传播算法和adadetla方法训练模型。将每个模型的迭代次数(nb_epoch)设置为100,将batch_size设为100,并在每个epoch之后验证模型。然后使用早停技巧来停止训练,因为有时错误率可能会到后期有所波动。基于验证阶段的准确性情况选择最佳的模型。
[0041]
两个模型实验效果比较如图5所示。
[0042]
本实施例记录了69组数据针对两类模型的auc曲线数据,并进行比较。如图5所示,新的模型使用将dna序列数据与dna形状数据相结合的深度学习整合框架来预测tfbs。实验评估表明,整合框架具有比基于初级dna序列的模型更好的性能和准确度(auc的值越高准确度相对越高)。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1