一种基于BP人工神经网络预测有机化学分子基态能量的方法
一种基于bp人工神经网络预测有机化学分子基态能量的方法
技术领域
1.本发明涉及计算机化学技术领域,具体涉及一种基于bp人工神经网络预测有机化学分子基态能量的方法。
背景技术:
2.基态能量是分子的基本属性,计算有机化学分子基态能量为分析和筛选化合物和材料提供了数据支持,加速了发现新分子和设计新材料的进程。尽管在数据库中有大量的对外开放的数据支持,但对于没有发现的新分子新材料的数据,仍然需要使用电子结构理论进行大量的模拟计算,其中比较流行的计算方法是基于单粒子轨道kohn
‑
sham方程的密度泛函理论,但是这种方法存在如下问题:
3.1)计算成本高,计算成本也会根据分子中原子的数量而呈现指数级增长;
4.2)计算非常耗时,根据分子中原子的数量,整个计算过程非常复杂,需要耗费较长时间;
5.3)预测准确率低且其泛化能力和鲁棒性均较低。
技术实现要素:
6.发明目的:针对现有技术中存在的问题,本发明提供一种基于bp人工神经网络预测有机化学分子基态能量的方法,使用分子库伦矩阵作为特征输入,利用训练集对bp人工神经网络模型进行训练,得到稳定最优的基态能量预测模型,使用训练后的模型预测分子基态能量,实现高效准确的基态能量预测。
7.技术方案:本发明提供了一种基于bp人工神经网络预测有机化学分子基态能量的方法,包括如下步骤:
8.步骤1:收集有机化学分子信息,构建电子结构数据库;
9.步骤2:根据所述电子结构数据库,筛选出满足条件的分子信息;
10.步骤3:根据所述筛选出的分子信息整理出gjf输入文件,并计算分子基态能量;
11.步骤4:使用电子结构数据库计算表示分子中原子的笛卡尔坐标和核电荷的库伦矩阵,对分子库伦矩阵进行特征值计算,并输出一维库伦矩阵向量,将其作为bp人工神经网络的特征输入;
12.步骤5:对特征输入进行数据预处理,分析特征输入与基态能量之间的关联性,并整理数据预测的csv输入文件,其包含了分子的物质识别号、一维库伦矩阵向量、分子基态能量,所述csv输入文件分为训练数据和测试数据;
13.步骤6:构建bp人工神经网络模型预测有机化学分子基态能量,通过训练数据训练得到最优模型构型和超参数大小;
14.步骤7:将测试数据输入所述bp神经网络模型中训练,通过评价模型预测结果。
15.进一步地,所述步骤2筛选分子信息需要满足的条件为:
16.1)每个分子必须由c、h、o、n、p、s集合或其子集元素组成;
17.2)每个分子必须最少有2个原子,最多有50个原子;
18.3)为了平面波计算的收敛,分子中两个原子之间的最大距离不得超过25a0,其中a0=5.29
×
10
‑
11
m,即玻尔半径,每个分子被放置在边长为30a0的立方盒中;
19.4)分子中必须有偶数个电子。
20.3.根据权利要求1所述的基于bp人工神经网络预测有机化学分子基态能量的方法,其特征在于,所述步骤3分子基态能量通过optb3lyp/6
‑
31g方法计算。
21.进一步地,所述库伦矩阵的计算公式如下:
[0022][0023]
其中,i,j表示矩阵中非对角元素对应的原子,z
i
表示原子序数,r
i
表示原子坐标,非对角元素表示原子i和j之间库仑排斥,对角元素表示由原子序数拟合核电荷得到的原子能量。
[0024]
进一步地,设定所述库伦矩阵大小为50
×
50,分子中原子数量少于50个的,剩下部分使用0进行填充,所述分子库伦矩阵特征值为:
[0025]
λ=
x
‑
l
cx
[0026]
其中,c为库伦矩阵,所述库伦矩阵通过处理后变为λ
i
,i=50的一维库伦矩阵向量。
[0027]
进一步地,所述步骤5中的预处理包括:
[0028]
步骤5.1:将所述一维库伦矩阵向量使用pca算法将库伦矩阵由50维数据降维成2维数据,具体为:
[0029]
1)将原始数据按列组成n行d列矩阵x;
[0030]
2)将x的每一列进行零均值化,即x
‑
x
mean
;
[0031]
3)求出协方差矩阵其中,m表示矩阵维度,此处等于50;
[0032]
4)求出协方差矩阵的特征值及对应的特征向量;
[0033]
5)将特征向量按对应特征值大小从上到下按行排列成矩阵,取前2行组成矩阵p;
[0034]
6)y=px即为降维到2维后的数据。
[0035]
步骤5.2将降维后的一维库伦矩阵向量进行归一化操作,所述归一化处理使用的是(0,1)标准差公式;
[0036]
进一步地,所述步骤6具体操作为:
[0037]
步骤6.1:所述bp人工神经网络采用四层神经层,其中包含了两层隐藏层,激活函数选择relu函数,损失函数选择均方差损失函数,并且设置学习率(α)初始值为0.003,监听损失函数的值,当损失函数值增长超过五次,则减少学习率;
[0038]
步骤6.2:使用adam优化算法进行参数和权重的更新,实现按照误差逆向传播算法训练的多层前馈神经网络,利用梯度的一阶矩估计和二阶矩估计动态调整每个参数的学习率;
[0039]
步骤6.3:将训练数据输入到构建出的bp人工神经网络中进行训练学习,将模型迭
代次数设置为100,直到模型最优稳定。
[0040]
进一步地,所述步骤7具体操作为:
[0041]
步骤7.1:将预测的结果和真实数据放在一个矩阵中进行对比,查看偏差大小;
[0042]
步骤7.2:所述评价模型使用均方差损失(mse)函数、决定系数(r2)以及校正决定系数(r2_adjusted)作为评价指标。
[0043]
有益效果:
[0044]
1、本发明bp人工神经网络通过算法对指定的分子数据库信息进行训练学习,bp人工神经网络是按照误差逆向传播算法训练的多层前馈神经网络,具有并行处理数据、自适应学习、误差反向传播等特点,预测出一组新分子的性质,预测准确率高,不需要进行新的模拟计算,节约了计算成本。
[0045]
2、本发明将分子的库伦矩阵作为特征输入,通过构建bp人工神经网络对训练数据进行学习,无需经验公式和数学模型,自动寻找库伦矩阵与基态能量之间复杂的非线性对应关系,并且具有较高的准确率,可以实现基于分子库伦矩阵准确地预测出分子基态能量。
[0046]
3、本发明使用bp人工神经网络预测分子基态能量,与dft计算相比,计算时间不会根据分子中原子的数量而呈现指数级增长,而是一直保持在亚秒速度,计算成本低。
[0047]
4、本发明使用bp人工神经网络预测分子基态能量,训练出的模型可以快速准确地预测新分子的基态能量,具有较好的泛化能力和较强的鲁棒性。使用bp人工神经网络预测有机化学分子基态能量为分析和筛选化合物和材料提供了数据支持,加速了发现新分子和设计新材料的进程。
附图说明
[0048]
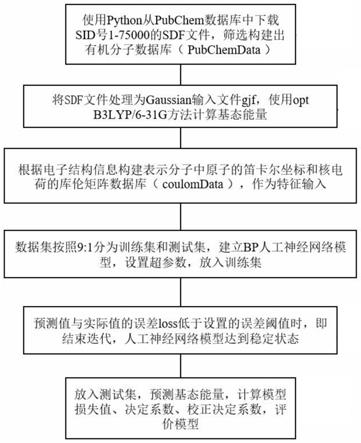
图1为本发明预测方法流程图;
[0049]
图2为本发明基态能量分布的质量分布图;
[0050]
图3为本发明乙烯结构与库伦矩阵图;
[0051]
图4为本发明降维特征和基态能量关系散点图;
[0052]
图5为本发明预测基态能量的bp神经网络模型结构图;
[0053]
图6为本发明bp神经网络模型训练误差曲线图。
具体实施方式
[0054]
下面结合附图对本发明作进一步描述。以下实施例仅用于更加清楚地说明本发明的技术方案,而不能以此来限制本发明的保护范围。
[0055]
如图1所示,本发明公开了一种基于bp人工神经网络预测有机化学分子基态能量的方法,其具体步骤如下:
[0056]
步骤1:首先要收集有机化学分子信息,构建出电子结构数据库,根据电子结构数据库整理出gjf输入文件,计算基态能量。
[0057]
步骤1.1:收集分子信息的方式使用pubchem数据库提供的pubchempy科学库接口,从数据库中收集物质识别号在1到75000的分子信息,根据数据库提供的分子结构信息,并筛选出满足以下条件的分子信息:
[0058]
1)每个分子必须由c、h、o、n、p、s集合或其子集元素组成;
[0059]
2)每个分子必须最少有2个原子,最多有50个原子;
[0060]
3)为了平面波计算的收敛,分子中两个原子之间的最大距离不得超过25a0(a0=5.29
×
10
‑
11
m,即玻尔半径),每个分子被放置在边长为30a0的立方盒中;
[0061]
4)分子中必须有偶数个电子。
[0062]
步骤1.2:筛选出的分子结构数据文件(sdf)被转换为电子结构计算的输入文件(gjf),输入gaussian程序,使用optb3lyp/6
‑
31g方法计算基态能量,分子基态能量分布图如图2所示,分子基态能量大多数分布在[
‑
1000,0]范围内。
[0063]
表1部分分子基态能量
[0064][0065][0066]
表2基态能量的数值统计
[0067][0068]
从上表中可以看出基态能量的最大值和最小值的差值较大,很容易导致损失函数值较大,需要画出降维后的特征输入和基态能量之间的函数散点图判断是否存在很强的关联关系。
[0069]
步骤2:为提高预测精度并减少计算量,将使用库伦矩阵作为机器学习预测的输入。
[0070]
步骤2.1:库伦矩阵表示分子中原子的笛卡尔坐标和核电荷的矩阵,包含原子核的排斥和自由原子的势能信息,并且对分子的平移和旋转保持不变,将分子信息使用数据矩
阵的方式表示出来,库伦矩阵的计算公式如下所示。
[0071][0072]
其中,i,j表示矩阵中非对角元素对应的原子,z
i
表示原子序数,r
i
表示原子坐标,其中,非对角元素表示原子i和j之间库仑排斥,对角元素表示由原子序数拟合核电荷得到的原子能量。如图2所示是将乙烯使用库伦矩阵表示。
[0073]
步骤2.2:由图2可以看出分子的一些关键信息,如分子中原子的笛卡尔坐标和原子序数都是用数组矩阵的方式表示出来。但使用库伦矩阵来表示分子信息的方法并不唯一,因为一个指定的分子可以由多个矩阵表示出来,通过重新调整库伦矩阵中原子的位置就可以获取到不同的库伦矩阵,这些矩阵之间的不同点就是行变换或列变换。
[0074]
表示方法不唯一就会导致机器学习的模型泛化能力较差,预测精度不高,所以为解决库伦矩阵表示不唯一问题,引入了库伦矩阵的特征值表示法,由图2可以看出库伦矩阵一定是一个实对称矩阵,根据实对称矩阵一定有特征值且特征值为实数的性质,计算出库伦矩阵对应的特征值,由此可以将给定的分子信息的库伦矩阵表示成将特征值由大到小排列的唯一一维向量(λ1,λ2,
…
,λ
n
),λ
i
≥λ
i+1
,并且为了保持特征值的数量一致,需要固定库伦矩阵的大小。
[0075]
步骤2.3:在收集分子数据信息阶段规定了分子中原子的数量不能超过50个,可以将分子信息构建出的库伦矩阵规定为50
×
50的矩阵,若分子中原子数量少于50个剩下部分使用0进行填充,由于50
×
50库伦矩阵是一个实对称矩阵,为了节省存储空间,将只存储矩阵主对角线的上三角,将存储量由原先一个分子库伦矩阵使用2500个数字组成缩减为一个分子库伦矩阵使用1275个数字组成,之后求出50
×
50库伦矩阵的50个特征值从小到大存储到一维向量中,计算特征值公式如下所示。
[0076]
λ=x
‑
l
cx
[0077]
以乙烯为例,处理后的库伦矩阵和特征值如下所示。
[0078][0079]
库伦矩阵通过处理后变为了λ
i
,i≠50的一维向量,然后将计算出的50个特征值组成的向量作为机器学习的输入,预测出分子的基态能量,机器学习的任务就是找出一个非线性函数,反映出输入的特征值向量和基态能量之间的关系。
[0080]
步骤2.4:将上述方法应用到所有的数据中,数据量一共是16241条,计算出的库伦矩阵结果如下所示。
[0081]
表3分子库伦矩阵
[0082][0083][0084]
对分子的库伦矩阵进行特征值的计算,计算结果如下所示:
[0085]
表4分子库伦矩阵特征值
[0086][0087]
计算出的分子库伦矩阵特征值的形状为(16241,50),将会被作为bp神经网络的特征输入。
[0088]
步骤2.5:将计算出的分子库伦矩阵特征值进行数值分析。
[0089]
表5库伦矩阵特征值数值分析
[0090]
[0091][0092]
根据上表库伦矩阵特征值数值统计可以看出,最大值和最小值之间差值较大,后续还需要进行归一化处理。
[0093]
步骤3:分析特征输入与基态能量之间的关联性,并进行数据预处理。
[0094]
步骤3.1:数据集中一共有16241条数据,最终存储库伦矩阵的一维向量形状为(16241,50)设为矩阵x,使用pca算法将库伦矩阵降维成(16241,2),提取出数据的主要特征分量。主要步骤如下:
[0095]
1)将原始数据按列组成n行d列矩阵x;
[0096]
2)将x的每一列(代表一个属性)进行零均值化,即x
‑
x
mean
;
[0097]
3)求出协方差矩阵其中m表示矩阵维度,此处等于50;;
[0098]
4)求出协方差矩阵的特征值及对应的特征向量;
[0099]
5)将特征向量按对应特征值大小从上到下按行排列成矩阵,取前2行组成矩阵p;
[0100]
6)y=px即为降维到2维后的数据。
[0101]
输出降维到2维的数据如下所示。
[0102]
表5部分二维库伦矩阵
[0103][0104][0105]
画出降维的二维库伦矩阵和对应基态能量之间的散点图如图3所示,发现选择的特征与基态能量之间具有很强的规律性。
[0106]
步骤3.2:接下来整理数据预测的csv输入文件,包含了分子的物质识别号、一维库伦矩阵向量、分子基态能量,其中作为特征输入的特征矩阵将被归一化处理,使用的是(0,
1)标准差公式为:
[0107][0108]
其中,x表示输入的特征矩阵,形状为(16241,50)。
[0109]
构建出的csv输入文件行列的形状为(16241,52),包含了16241个分子信息,分子数据90%会被划分为训练数据,剩下的10%当做测试数据。
[0110]
步骤4:构建bp人工神经网络预测有机化学分子基态能量,通过训练数据训练得到最优模型构型和超参数大小。
[0111]
步骤4.1:经过大量的训练,最终建立的bp人工神经网络采用四层神经层,其中包含了两层隐藏层,神经网络一共需要训练54273个参数,激活函数选择relu函数,增加模型非线性,修正线性单元(relu)公式如下:
[0112][0113]
损失函数选择均方差损失(mse)函数:
[0114][0115]
并且设置学习率(α)初始值为0.003,监听损失函数的值,当损失函数值增长超过五次,则减少学习率。
[0116]
优化算法选择adam算法,使用adam进行参数和权重的更新,实现按照误差逆向传播算法训练的多层前馈神经网络,目的是使损失函数值达到最小,利用梯度的一阶矩估计和二阶矩估计动态调整每个参数的学习率,adam算法的优点主要在于经过偏置校正后,每一次迭代学习率都有个确定范围,使得参数比较平稳。公式如下:
[0117]
m
t
=μ*m
t
‑1+(1
‑
μ)*g
t
[0118][0119][0120][0121][0122]
其中,m
t
,n
t
分别是对梯度的一阶矩估计和二阶矩估计,可以看作对期望e|g
t
|,的估计,是对m
t
,n
t
的校正,这样可以近似为对期望的无偏估计,可以看出,直接对梯度的矩估计对内存没有额外的要求,而且可以根据梯度进行动态调整,而对学习率形成一个动态约束,而且有明确的范围。
[0123]
步骤4.2:构建出bp人工神经网络的结果如下所示。
[0124]
表6网络模型结构表
[0125][0126]
从表中可以看出,bp人工神经网络模型结构包括了四层结构,其中隐藏层有两层,需要训练的参数数量为54273个,经过多层神经网络,最终输出预测的分子基态能量值。
[0127]
步骤4.3:将训练数据集放入到构建出的bp人工神经网络中进行训练学习,将模型迭代次数设置为100,直到模型最优稳定,输出的损失函数值被记录下来,最终模型损失函数值在0.5457时模型最优。
[0128]
步骤5:将测试数据放入构建出的bp神经网络模型中训练,而且评价模型预测结果。
[0129]
步骤5.1:将预测的结果和真实数据放在一个矩阵中进行对比,查看偏差大小。
[0130]
表7部分真实值和预测值对比表
[0131][0132][0133]
从表中可以看出,模型预测出的基态能量值和真实值之间的误差非常小,表明模型预测的准确率非常高,但表中显示的只是部分数据,还需要使用确定系数和校正决定系数查看模型总体的表现。
[0134]
步骤5.2:模型预测的评价标准使用了均方差损失(mse)函数:
[0135][0136]
模型预测测试集数据的均方差损失值为0.7592,与模型学习训练集的均方差0.5457差值不大,模型表现非常好,具有很强的鲁棒性。
[0137]
步骤5.2:模型拟合的评价指标还采用了决定系数(r2)作为模型拟合的评价标准,
r2算法计算公式为:
[0138][0139]
其中,y,分别是真实值和预测值,取值范围为[0,1],越接近1表示模型拟合越好,训练好的模型使用测试集进行测试得到的值为0.999,结果显示模型拟合非常好。
[0140]
步骤5.3:模型预测的评价指标采用了校正决定系数(r2_adjusted)作为模型预测的评价标准,r2_adjusted算法计算公式为:
[0141][0142]
其中,参数r2,n,p分别代表决定系数,样本数量,特征数量,r2_adjusted取值范围为[0,1],越接近1表示模型预测结果越精确,训练好的模型使用测试集进行测试得到的r2_adjusted值为0.999,结果显示模型预测结果非常精确。
[0143]
通过对16241条带标签的分子基态能量数据的处理,使用分子库伦矩阵作为特征输入,构建bp人工神经网络预测有机化学分子基态能量的方法,经过结果验证可知,预测精度可以达到99.998%,非常接近真实值,计算成本也非常低。
[0144]
上述实施方式只为说明本发明的技术构思及特点,其目的在于让熟悉此项技术的人能够了解本发明的内容并据以实施,并不能以此限制本发明的保护范围。凡根据本发明精神实质所做的等效变换或修饰,都应涵盖在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1