一种基于深度学习预测的器官芯片药物评价方法
1.本发明属于生物医学工程和计算机科学医工融合领域,设计了一种基于深度学习的器官芯片实验结果评价和预测方法。本方法可以用于多种不同类型的器官芯片药物治疗评价之中,对于单个器官芯片数据库,可以经过数据库中的已经带有标签数据的训练数据集对深度学习模型进行训练,进而预测加载药物以后器官芯片反应的实验结果。有效的解决了器官芯片与人工智能相结合的关键环节——如何利用器官芯片实验产生的大数据进行深度学习进而预测实验结果,用于药物治疗评价。
2.深度学习模型训练中用到的器官芯片实验相关的数据字段有不同种类,包括生物支架材料、试剂、细胞系、药物、器官芯片型号、器官芯片配置参数(试剂和药物浓度、细胞类型等),以及作为标签数据的时间信息和实验结果(细胞代谢物浓度、细胞的数量和存活率、芯片内微环境的ph、温度、氧气浓度、二氧化碳浓度、teer、气压、是否加入药物、药物的释放速度、降解速率),经过这些数据的训练可以得到深度学习模型权重矩阵,当输入如上的这些信息后模型会自动预测出带有时间维度的实验结果标签数据。
3.在建立深度学习模型之前,需要对如上提到的器官芯片相关数据进行预处理,需要把这些实际数据(文本的和非文本的信息,诸如生物支架材料和药物试剂的名称、分子式、成份等)转换成深度学习模型可以理解和计算的数字信息,这里可以用到的数据转换技术包括mol2vec、word2vec、gene2vec、one-hot编码表示等算法工具。
背景技术:
4.药物评价是药物上市前获得fda批准的重要原因之一,因此在新药物开发过程中,以及在成品药治疗新症状的筛选过程中,药物评价是至关重要的环节,包括安全性、有效性、代谢能力、经济性、质量等,这些都属于药物治疗评价范畴,简称therapy assessment。这些评价指标对新药上市的进程有着很大的影响。由于动物模型具有整体性,其广泛的应用于临床前的毒性评价。然而,由于动物模型和人体模型在微环境上有着显著的物种差异,因此经常会导致药物的毒性、有效性、代谢能力等的实际临床结果和模型预测结果存在很大的偏差,导致药物研发周期变长变慢,大大提高了研发成本。目前,器官芯片被证明是用于临床前药物评价的非常有意义的体外模型,它可以加快药物发现和药物筛选的研发速度,缩短研发周期。同时在精准医疗领域,器官芯片依然发挥着重要作用。器官芯片可以避免或者减少动物实验次数,用自体细胞搭建肿瘤模型实现药物临床前测试和筛选。
5.器官芯片指的是一种在芯片上构建的器官生理微系统,它以微流控芯片为核心,通过与细胞生物学、生物材料和工程学等多种方法相结合,可以在体外模拟构建包含有多种活体细胞、功能组织界面、生物流体和机械力刺激等复杂因素的组织器官微环境,反映人体组织器官的主要结构和功能特征。这种组织器官模型不仅可在体外接近真实地重现人体器官的生理、病理活动,还可能使研究人员以前所未有的方式来见证和研究机体的各种生物学行为,预测人体对药物或外界不同刺激产生的反应,在生命科学研究、疾病模拟和新药研发等领域具有广泛应用价值。
6.器官芯片在培养和在其进行实验的过程中,会产生大量的实验数据,但是在以前的研究中,研究者们并没有仔细分析数据之间的关联,特别是对不同的器官芯片实验之间,数据并没有实现共享,所以也无法关注不同实验间的数据关联特点,只是一味看重实验结果,对实验过程中的数据,特别是动态数据丢失了,并且他们都只关心自己的实验数据,而没有时间和精力,也没有工具去关注别人曾经做过的类似实验结果,把自己的设计参数和其他人进行对比,因此,需要开发适当的数据分析方法来对这些数据进行分析和建模。
技术实现要素:
7.发明目的:
8.针对上述研究的问题,本发明的目的在于提供一种基于深度学习的器官芯片实验结果预测方法,结合深度学习算法,经过来自网络的开源数据库、实验室数据以及根据某种算法自动生成的模拟数据对深度学习模型进行训练,对实验结果进行预测,从而帮助药物研发人员更好的优化器官芯片参数配置,更好的调整药物设计方案,从而缩短新药物上市时间和精准药物在临床上使用之前的成品药物筛选时间。
9.实际上,我们是希望用深度学习的网络模型将药物、靶蛋白、试剂、支架材料的分子式和空间结构式这些微观信息,细胞的基因信息,以及它们之间的不同浓度比例等进行数据融合,在网络模型内部识别它们之间的关系,从而用于预测实验结果数据。
10.为了达到上述目的,本发明设计了一种基于深度学习预测的多种器官芯片药物评价方法,内容如下:
11.步骤1:建立器官芯片数据库,存储着与器官芯片实验相关的各种数据,包括生物支架材料数据表、试剂数据表、细胞系数据表、药物数据表、器官芯片型号数据表、器官芯片参数配置数据表(试剂和药物浓度、细胞类型等),以及可以作为标签数据的带有时间信息的实验结果数据表(细胞代谢物浓度、细胞的数量和存活率、芯片内微环境的ph、温度、氧气浓度、二氧化碳浓度、teer、气压、是否加入药物、药物的释放速度、降解速率)等。
12.步骤2:向器官芯片数据库支架材料表、细胞系表、药物表、试剂表中导入网络开源信息作为基础数据,例如,材料相关的matweb、matmatch网络数据库,细胞系相关的panglaodb、cellmarker网络数据库,试剂相关的thermofisher数据库,药物信息相关的pubchem、drugbank数据库;再向器官芯片型号数据表中导入目前市场上已知的器官芯片产品信息,以及一些知名期刊报道的科研团队作品和数据;再向器官芯片参数配置表中导入来自于知名期刊科研论文或者研究机构实验室数据的器官芯片配置信息,即在器官芯片实验中所用到的实验数据;再向带有时间维度的实验结果数据表中导入期刊文章的实验结果数据或者实验室采集到的测试结果数据。
13.此外,还可以用基于知识图谱的方法自动生成部分模拟数据导入到器官芯片参数配置表和带有时间维度的实验结果表,这样的模拟数据其实质是让模型学习到人类的经验规则,即知识图谱。
14.需要注意的是,如果实验结果中涉及到的数值类的数据很少,可以将其转化为等级分类的数据类型,例如,氧气含量<19.5%为等级1;19.5%<氧气含量<24%为等级2;氧气含量>24%为等级3,便于深度学习模型计算。
15.从药物的角度,可以把fda批准的药物产生的相关数据作为正样本数据集,未经批
准的药物作为负样本数据集,对于网络数据库而言大部分数据都是正样本,因为模型中带有分子式等微观结构信息,所以用正样本数据预测负样本数据的实验结果是可行的。但如果是二分类的药物安全性评价,由于训练数据集都是正样本造成数据的过拟合,导致无法预测负样本未经fda批准药物数据的安全性二分类结果。
16.步骤3:将器官芯片数据库中的数据进行数据预处理以及向量化的特征表示。
17.步骤3.1:数据的预处理:将器官芯片数据库所有字段数据进行预处理,其中主要包括对缺失值进行填补和对不准确、不相关的数据进行替换或删除,以提高数据集质量,保证不同数据源数据格式的统一。
18.步骤3.2:特征向量化:器官芯片数据库中存放着大量数据,其中部分数据是可以被深度学习模型所运用进行计算的,这部分数据在输入到模型之前需要进行向量化表示,即将这些文本或者非文本数据转换成数字向量编码表示,便于模型计算。向量化的方法如下:
19.(1)对于药物信息相关的数据表(存储着药物名称、分子式、二维和三维结构式、靶蛋白、smile格式表达式、mol2vec编码等,其中靶蛋白信息需要建立蛋白质数据表和靶点数据表来表述drug target interaction,简称dti关联信息),药物分子式可以使用摩根算法转为指纹信息,由于指纹信息数字位数过长,所以可以再经过某种模型进行二次训练转换,比如可以经过bert算法再输出向量,或者药物分子式也可以经过mol2vec算法直接转换为向量,转换后的数字字符串结果可以直接存储在药物信息表中。对于靶蛋白质氨基酸序列的向量化,可以把氨基酸序列用pssm方法表示,其余信息,如果是数字可以使用0到1之间的归一化方法编码,如果是文本可以使用one-hot编码。
20.pssm矩阵表示方法如下:
21.第一步,首先找到某个蛋白质fasta序列以及同源蛋白质fasta序列,将他们按行(列)进行排列;第二步,然后计算每个序列的每种氨基酸的个数,得到ppm矩阵,是一个l*20的矩阵,20代表人体的氨基酸的个数,l代表蛋白质序列的长度;第三步,然后对矩阵标准化得到pfm矩阵;第四步,然后根据公式求得pssm矩阵,pssm矩阵是一个l*20的矩阵,20代表人体的氨基酸的个数,l代表蛋白质序列的长度。该矩阵既可以表示某个蛋白质也表示每个位置上氨基酸突变成为其他氨基酸的可能。每行中最大的数字对应的氨基酸,就是这个矩阵代表的蛋白质。每个元素代表了该位置的氨基酸突变成其他氨基酸的可能性。元素值越大,越可能发生突变。
22.(2)对于细胞信息相关的数据表(存储着细胞系名称、来源、基因序列、gene2vec编码等),细胞基因序列可以用gene2vec方法进行向量化,并存入细胞信息数据表。其余信息,如果是数字可以使用0到1之间的归一化方法编码,如果是文本可以使用one-hot编码。
23.(3)对于生物支架材料信息相关的数据表(存储着分子式、结构式、编码表示等),分子式和结构式可以用mol2vec方法进行向量化,并存入支架材料信息数据表。其余信息,如果是数字可以使用0到1之间的归一化方法编码,如果是文本可以使用word2vec或者one-hot编码。
24.(4)对于生物试剂信息相关的数据表(存储着成份、比例、浓度、化学式、结构式、编码等),化学式可以用mol2vec方法进行向量化,并存入试剂信息数据表。其余信息,如果是数字可以使用0到1之间的归一化方法编码,如果是文本可以使用word2vec或者one-hot编
码。
25.(5)对于器官芯片型号相关的数据表(存储着芯片型号id、枚举型变量的器官芯片种类、开发者、机构、文章名称及链接、官网介绍链接、芯片结构和组件描述、工作原理描述、word2vec编码等),这些字段信息,一部分数据比如官网链接、开发者、文章名称等和实验结果预测无关,因为不需要输入到人工智能模型中所以不需要向量化。其余信息,如果是数字可以使用0到1之间的归一化方法编码,如果是文本可以使用word2vec编码。
26.(6)对于器官芯片参数配置数据表(存储着参数配置id,便于和实验数据表关联,一行参数配置id信息对应着带有时间数据的多行实验数据表信息,还存储着器官芯片型号id,一种或者几种药物调配信息,生物试剂调配信息,所采用的支架材料调配信息,采用了哪几种细胞系等),如果是数字可以使用0到1之间的归一化方法编码,如果是文本可以使用word2vec或者one-hot编码。
27.(7)对于带有时间信息的实验结果数据表(存储着细胞代谢物浓度、细胞的数量和存活率、芯片内微环境的ph、温度、氧气浓度、二氧化碳浓度、teer、气压、是否加入药物、药物的释放速度、降解速率等),如果是数字可以使用0到1之间的归一化方法编码,如果是文本可以使用word2vec或者one-hot编码。如果用于训练模型的数据集数量很少,建议将实验结果中涉及到的数值类的数据转化为等级分类的数据类型,例如,氧气含量<19.5%为等级1;19.5%<氧气含量<24%为等级2;氧气含量>24%为等级3。
28.步骤4:深度学习模型的设计。数据向量化以后可以用于输入深度学习模型,而对于深度学习模型的设计,方案如下。
29.对于器官芯片数据库中数据的向量化,最终产生的向量都是数字字符串,把这些数字字符串连接起来长度是非常庞大的,而这个数字字符串每一位都需要对应深度学习模型输入层的一个输入端口,可见,端口数量也是非常庞大的。为了减少之后神经网络运算量节省算力,有必要对这个带有巨大端口数量的输入层进行数据压缩再交给网络中的神经元系统进行计算。
30.(1)对于药物信息相关数据表中的数据向量,smiles格式的药物分子字符串和蛋白质的氨基酸序列都是序列数据(smiles结构式中字符的顺序是遵循一定的规则的,如果互换的话,结构就不一样了,而且有同分异构体,分子式相同结构不同,但smiles字符串的表达是不同的),这些序列数据如果位数很长的话可以通过循环神经网络的lstm或者gru方法,或者卷积神经网络cnn进行关键特征识别进而压缩这些向量的长度,有助于减少后续全连接网络的计算量。此表中如果有较长的one-hot或者word2vec向量编码,也可以考虑使用自编码器(autoencoder)或者机器学习主成分分析算法(principal component analysis,pca),一种有监督的(supervised)线性降维算法(lda,linear discriminant analysis),局部线性嵌入算法(locally linear embedding,lle),拉普拉斯特征映射算法(laplacian eigenmaps),或者其他方法进行降维。其余向量,因为数字位长度比较短,所以可以不经过任何压缩或者关键特征识别,直接连接到全连接网络的输入层。
31.(2)对于细胞信息相关数据表中的数据向量,gene2vec是序列数据,如果位数很长的话可以通过循环神经网络的lstm或者gru方法,或者卷积神经网络cnn进行关键特征识别进而压缩这些向量的长度,有助于减少后续全连接网络的计算量。此表中如果有较长的one-hot或者word2vec向量编码,也可以考虑使用自编码器(autoencoder)或者机器学习主
linear embedding,lle),拉普拉斯特征映射算法(laplacian eigenmaps),或者其他方法进行降维。其余向量,因为数字位长度比较短,所以可以不经过任何压缩或者关键特征识别,直接连接到全连接网络的输入层。
36.(7)对于带有时间信息的实验结果数据表中的数据向量,都是数字型信息,时间格式也可以用相对表示法转换成数字,不存在序列数据。此表中如果有较长的one-hot或者word2vec向量编码,也可以考虑使用自编码器(autoencoder)或者机器学习主成分分析算法(principal component analysis,pca),一种有监督的(supervised)线性降维算法(lda,linear discriminant analysis),局部线性嵌入算法(locally linear embedding,lle),拉普拉斯特征映射算法(laplacian eigenmaps),或者其他方法进行降维。其余向量,因为数字位长度比较短,所以可以不经过压缩或者关键特征识别,直接连接到全连接网络的输入层。
37.对于使用卷积网络cnn对序列化数据进行有效特征提取和压缩,和lstm一样都是在全连接网络前对输入数据的一种数据压缩降维。卷积网络是把输入的字符串数据转换成行列矩阵,再经过设计若干个卷积核生成卷积层,再取最大值或者平均值进行池化,然后再拉平变成一维数组接入全连接网络。
38.总而言之,模型输入层因涉及到特征比较多,故输入的样本数据有着较高的维度,因此需要进行降维处理,然后将降维后的数据输入到全连接神经网络的输入层,降维后的一条数据中的每一个值/位对应输入层的每一个神经元,经过若干个隐藏层后,由输出层输出最终多维度的器官芯片实验结果,这个实验结果数据和带有时间信息的实验结果数据表中每一列数据一一对应。
39.步骤5:深度学习模型的训练及评估。
40.步骤5.1:对于输入样本x与输出样本y组成的数据集,按照一定的比例,将其划分为训练集与验证集,同时选取适当的代价函数和深度学习模型优化器,使用训练数据集对模型中的权值和偏置进行训练,以达到输出结果与实际结果误差逐渐减小到可接受的水平。在使用训练数据集对模型训练完成之后,需要使用验证集,对模型的超参数(神经网络中的神经元数和层数等)进行调整,以及使用正则化的方法,防止模型的过拟合,最终得到训练好的器官芯片药物评价模型。
41.步骤5.2:加载已经训练好的深度学习模型,将测试数据集中的数据作为模型输入,模型的输出即为对器官芯片实验结果的预测。
42.本发明优点在于:
43.(1)很好的将深度学习模型和器官芯片药物评价预测方法结合起来,具有了一定的智能特征。
44.(2)采用了多种对器官芯片数据进行向量化的方法,包括摩根算法、mol2vec、word2vec、pssm等。
45.(3)采用了多种对器官芯片数据向量进行降维和有效特征提取的方法,包括循环神经网络lstm和gru,卷积神经网络,主成分分析等,减少了全连接网络计算的压力。
46.(4)在深度学习模型中加入了时间维度数据,实现了对不同时间节点的实验结果预测。
47.(5)使用深度学习的方法,在面对大量的以及多维度的数据时有着强大的运算预
测能力。
附图说明
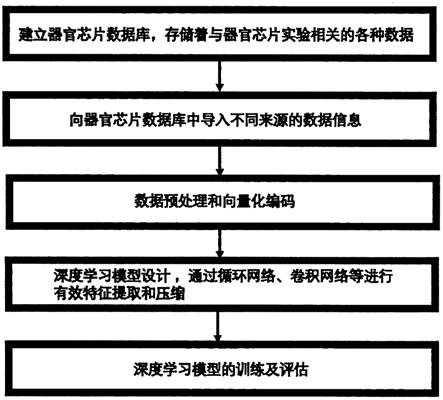
48.图1为基于深度学习预测的器官芯片药物评价方法的流程图;
具体实施方式
49.下面结合附图对本发明做出进一步的说明:
50.请参照图1,图1是本发明一种基于深度学习预测的器官芯片药物评价方法总体的流程图,总的流程包括如下步骤:
51.步骤1:建立器官芯片数据库,存储着与器官芯片实验相关的各种数据,包括生物支架材料数据表、试剂数据表、细胞系数据表、药物数据表、器官芯片型号数据表、器官芯片参数配置数据表(试剂和药物浓度、细胞类型等),以及可以作为标签数据的带有时间信息的实验结果数据表(细胞代谢物浓度、细胞的数量和存活率、芯片内微环境的ph、温度、氧气浓度、二氧化碳浓度、teer、气压、是否加入药物、药物的释放速度、降解速率)等。
52.步骤2:向器官芯片数据库支架材料表、细胞系表、药物表、试剂表中导入网络开源信息作为基础数据,例如,材料相关的matweb、matmatch网络数据库,细胞系相关的panglaodb、cellmarker网络数据库,试剂相关的thermofisher数据库,药物信息相关的pubchem、drugbank数据库;再向器官芯片型号数据表中导入目前市场上已知的器官芯片产品信息,以及一些知名期刊报道的科研团队作品和数据;再向器官芯片参数配置表中导入来自于知名期刊科研论文或者研究机构实验室数据的器官芯片配置信息,即在器官芯片实验中所用到的实验数据;再向带有时间维度的实验结果数据表中导入期刊文章的实验结果数据或者实验室采集到的测试结果数据。
53.此外,还可以用基于知识图谱的方法自动生成部分模拟数据导入到器官芯片参数配置表和带有时间维度的实验结果表,这样的模拟数据其实质是让模型学习到人类的经验规则,即知识图谱。
54.需要注意的是,如果实验结果中涉及到的数值类的数据很少,可以将其转化为等级分类的数据类型,例如,氧气含量<19.5%为等级1;19.5%<氧气含量<24%为等级2;氧气含量>24%为等级3,便于深度学习模型计算。
55.从药物的角度,可以把fda批准的药物产生的相关数据作为正样本数据集,未经批准的药物作为负样本数据集,对于网络数据库而言大部分数据都是正样本,因为模型中带有分子式等微观结构信息,所以用正样本数据预测负样本数据的实验结果是可行的。但如果是二分类的药物安全性评价,由于训练数据集都是正样本造成数据的过拟合,导致无法预测负样本未经fda批准药物数据的安全性二分类结果。
56.步骤3:将器官芯片数据库中的数据进行数据预处理以及向量化的特征表示。
57.步骤3.1:数据的预处理:将器官芯片数据库所有字段数据进行预处理,其中主要包括对缺失值进行填补和对不准确、不相关的数据进行替换或删除,以提高数据集质量,保证不同数据源数据格式的统一。
58.步骤3.2:特征向量化:器官芯片数据库中存放着大量数据,其中部分数据是可以被深度学习模型所运用进行计算的,这部分数据在输入到模型之前需要进行向量化表示,
即将这些文本或者非文本数据转换成数字向量编码表示,便于模型计算。向量化的方法如下:
59.(1)对于药物信息相关的数据表(存储着药物名称、分子式、二维和三维结构式、靶蛋白、smile格式表达式、mol2vec编码等,其中靶蛋白信息需要建立蛋白质数据表和靶点数据表来表述drug target interaction,简称dti关联信息),药物分子式可以使用摩根算法转为指纹信息,由于指纹信息数字位数过长,所以可以再经过某种模型进行二次训练转换,比如可以经过bert算法再输出向量,或者药物分子式也可以经过mol2vec算法直接转换为向量,转换后的数字字符串结果可以直接存储在药物信息表中。对于靶蛋白质氨基酸序列的向量化,可以把氨基酸序列用pssm方法表示,其余信息,如果是数字可以使用0到1之间的归一化方法编码,如果是文本可以使用one-hot编码。
60.pssm矩阵表示方法如下:
61.第一步,首先找到某个蛋白质fasta序列以及同源蛋白质fasta序列,将他们按行(列)进行排列;第二步,然后计算每个序列的每种氨基酸的个数,得到ppm矩阵,是一个l*20的矩阵,20代表人体的氨基酸的个数,l代表蛋白质序列的长度;第三步,然后对矩阵标准化得到pfm矩阵;第四步,然后根据公式求得pssm矩阵,pssm矩阵是一个l*20的矩阵,20代表人体的氨基酸的个数,l代表蛋白质序列的长度。该矩阵既可以表示某个蛋白质也表示每个位置上氨基酸突变成为其他氨基酸的可能。每行中最大的数字对应的氨基酸,就是这个矩阵代表的蛋白质。每个元素代表了该位置的氨基酸突变成其他氨基酸的可能性。元素值越大,越可能发生突变。
62.(2)对于细胞信息相关的数据表(存储着细胞系名称、来源、基因序列、gene2vec编码等),细胞基因序列可以用gene2vec方法进行向量化,并存入细胞信息数据表。其余信息,如果是数字可以使用0到1之间的归一化方法编码,如果是文本可以使用one-hot编码。
63.(3)对于生物支架材料信息相关的数据表(存储着分子式、结构式、编码表示等),分子式和结构式可以用mol2vec方法进行向量化,并存入支架材料信息数据表。其余信息,如果是数字可以使用0到1之间的归一化方法编码,如果是文本可以使用word2vec或者one-hot编码。
64.(4)对于生物试剂信息相关的数据表(存储着成份、比例、浓度、化学式、结构式、编码等),化学式可以用mol2vec方法进行向量化,并存入试剂信息数据表。其余信息,如果是数字可以使用0到1之间的归一化方法编码,如果是文本可以使用word2vec或者one-hot编码。
65.(5)对于器官芯片型号相关的数据表(存储着芯片型号id、枚举型变量的器官芯片种类、开发者、机构、文章名称及链接、官网介绍链接、芯片结构和组件描述、工作原理描述、word2vec编码等),这些字段信息,一部分数据比如官网链接、开发者、文章名称等和实验结果预测无关,因为不需要输入到人工智能模型中所以不需要向量化。其余信息,如果是数字可以使用0到1之间的归一化方法编码,如果是文本可以使用word2vec编码。
66.(6)对于器官芯片参数配置数据表(存储着参数配置id,便于和实验数据表关联,一行参数配置id信息对应着带有时间数据的多行实验数据表信息,还存储着器官芯片型号id,一种或者几种药物调配信息,生物试剂调配信息,所采用的支架材料调配信息,采用了哪几种细胞系等),如果是数字可以使用0到1之间的归一化方法编码,如果是文本可以使用
word2vec或者one-hot编码。
67.(7)对于带有时间信息的实验结果数据表(存储着细胞代谢物浓度、细胞的数量和存活率、芯片内微环境的ph、温度、氧气浓度、二氧化碳浓度、teer、气压、是否加入药物、药物的释放速度、降解速率等),如果是数字可以使用0到1之间的归一化方法编码,如果是文本可以使用word2vec或者one-hot编码。如果用于训练模型的数据集数量很少,建议将实验结果中涉及到的数值类的数据转化为等级分类的数据类型,例如,氧气含量<19.5%为等级1;19.5%<氧气含量<24%为等级2;氧气含量>24%为等级3。
68.步骤4:深度学习模型的设计。数据向量化以后可以用于输入深度学习模型,而对于深度学习模型的设计,方案如下。
69.对于器官芯片数据库中数据的向量化,最终产生的向量都是数字字符串,把这些数字字符串连接起来长度是非常庞大的,而这个数字字符串每一位都需要对应深度学习模型输入层的一个输入端口,可见,端口数量也是非常庞大的。为了减少之后神经网络运算量节省算力,有必要对这个带有巨大端口数量的输入层进行数据压缩再交给网络中的神经元系统进行计算。
70.(1)对于药物信息相关数据表中的数据向量,smiles格式的药物分子字符串和蛋白质的氨基酸序列都是序列数据(smiles结构式中字符的顺序是遵循一定的规则的,如果互换的话,结构就不一样了,而且有同分异构体,分子式相同结构不同,但smiles字符串的表达是不同的),这些序列数据如果位数很长的话可以通过循环神经网络的lstm或者gru方法,或者卷积神经网络cnn进行关键特征识别进而压缩这些向量的长度,有助于减少后续全连接网络的计算量。此表中如果有较长的one-hot或者word2vec向量编码,也可以考虑使用自编码器(autoencoder)或者机器学习主成分分析算法(principal component analysis,pca),一种有监督的(supervised)线性降维算法(lda,linear discriminant analysis),局部线性嵌入算法(locally linear embedding,lle),拉普拉斯特征映射算法(laplacian eigenmaps),或者其他方法进行降维。其余向量,因为数字位长度比较短,所以可以不经过任何压缩或者关键特征识别,直接连接到全连接网络的输入层。
71.(2)对于细胞信息相关数据表中的数据向量,gene2vec是序列数据,如果位数很长的话可以通过循环神经网络的lstm或者gru方法,或者卷积神经网络cnn进行关键特征识别进而压缩这些向量的长度,有助于减少后续全连接网络的计算量。此表中如果有较长的one-hot或者word2vec向量编码,也可以考虑使用自编码器(autoencoder)或者机器学习主成分分析算法(principal component analysis,pca),一种有监督的(supervised)线性降维算法(lda,linear discriminant analysis),局部线性嵌入算法(locally linear embedding,lle),拉普拉斯特征映射算法(laplacian eigenmaps),或者其他方法进行降维。其余向量,因为数字位长度比较短,所以可以不经过任何压缩或者关键特征识别,直接连接到全连接网络的输入层。
72.(3)对于生物支架材料信息相关数据表中的数据向量,分子式和结构式的字符串表达,例如smiles格式或者mol2vec方法的数字字符串表达式,都是是序列数据,如果位数很长的话可以通过循环神经网络的lstm或者gru方法,或者卷积神经网络cnn进行关键特征识别进而压缩这些向量的长度,有助于减少后续全连接网络的计算量。此表中如果有较长的one-hot或者word2vec向量编码,也可以考虑使用自编码器(autoencoder)或者机器学习
主成分分析算法(principal component analysis,pca),一种有监督的(supervised)线性降维算法(lda,linear discriminant analysis),局部线性嵌入算法(locally linear embedding,lle),拉普拉斯特征映射算法(laplacian eigenmaps),或者其他方法进行降维。其余向量,因为数字位长度比较短,所以可以不经过任何压缩或者关键特征识别,直接连接到全连接网络的输入层。
73.(4)对于生物试剂信息相关数据表中的数据向量,试剂化学式和结构式的字符串表达,例如smiles格式或者mol2vec方法的数字字符串表达式,都是是序列数据,如果位数很长的话可以通过循环神经网络的lstm或者gru方法,或者卷积神经网络cnn进行关键特征识别进而压缩这些向量的长度,有助于减少后续全连接网络的计算量。此表中如果有较长的one-hot或者word2vec向量编码,也可以考虑使用自编码器(autoencoder)或者机器学习主成分分析算法(principal component analysis,pca),一种有监督的(supervised)线性降维算法(lda,linear discriminant analysis),局部线性嵌入算法(locally linear embedding,lle),拉普拉斯特征映射算法(laplacian eigenmaps),或者其他方法进行降维。其余向量,因为数字位长度比较短,所以可以不经过任何压缩或者关键特征识别,直接连接到全连接网络的输入层。
74.(5)对于器官芯片型号相关数据表中的数据向量,存在对芯片结构和工作原理的文字描述,这部分信息使用word2vec方法转换为向量,也是序列数据,因为位数比较长所以可以通过循环神经网络的lstm或者gru方法,或者卷积神经网络cnn进行关键特征识别进而压缩这些向量的长度,有助于减少后续全连接网络的计算量。此表中如果有较长的one-hot或者word2vec向量编码,也可以考虑使用自编码器(autoencoder)或者机器学习主成分分析算法(principal component analysis,pca),一种有监督的(supervised)线性降维算法(lda,linear discriminant analysis),局部线性嵌入算法(locally linear embedding,lle),拉普拉斯特征映射算法(laplacian eigenmaps),或者其他方法进行降维。其余向量,因为数字位长度比较短,所以可以不经过任何压缩或者关键特征识别,直接连接到全连接网络的输入层。
75.(6)对于器官芯片参数配置数据表中的数据向量,都是非序列数据。此表中如果有较长的one-hot或者word2vec向量编码,也可以考虑使用自编码器(autoencoder)或者机器学习主成分分析算法(principal component analysis,pca),一种有监督的(supervised)线性降维算法(lda,linear discriminant analysis),局部线性嵌入算法(locally linear embedding,lle),拉普拉斯特征映射算法(laplacian eigenmaps),或者其他方法进行降维。其余向量,因为数字位长度比较短,所以可以不经过任何压缩或者关键特征识别,直接连接到全连接网络的输入层。
76.(7)对于带有时间信息的实验结果数据表中的数据向量,都是数字型信息,时间格式也可以用相对表示法转换成数字,不存在序列数据。此表中如果有较长的one-hot或者word2vec向量编码,也可以考虑使用自编码器(autoencoder)或者机器学习主成分分析算法(principal component analysis,pca),一种有监督的(supervised)线性降维算法(lda,linear discriminant analysis),局部线性嵌入算法(locally linear embedding,lle),拉普拉斯特征映射算法(laplacian eigenmaps),或者其他方法进行降维。其余向量,因为数字位长度比较短,所以可以不经过压缩或者关键特征识别,直接连接到全连接网络
的输入层。
77.对于使用卷积网络cnn对序列化数据进行有效特征提取和压缩,和lstm一样都是在全连接网络前对输入数据的一种数据压缩降维。卷积网络是把输入的字符串数据转换成行列矩阵,再经过设计若干个卷积核生成卷积层,再取最大值或者平均值进行池化,然后再拉平变成一维数组接入全连接网络。
78.总而言之,模型输入层因涉及到特征比较多,故输入的样本数据有着较高的维度,因此需要进行降维处理,然后将降维后的数据输入到全连接神经网络的输入层,降维后的一条数据中的每一个值/位对应输入层的每一个神经元,经过若干个隐藏层后,由输出层输出最终多维度的器官芯片实验结果,这个实验结果数据和带有时间信息的实验结果数据表中每一列数据一一对应。
79.步骤5:深度学习模型的训练及评估。
80.步骤5.1:对于输入样本x与输出样本y组成的数据集,按照一定的比例,将其划分为训练集与验证集,同时选取适当的代价函数和深度学习模型优化器,使用训练数据集对模型中的权值和偏置进行训练,以达到输出结果与实际结果误差逐渐减小到可接受的水平。在使用训练数据集对模型训练完成之后,需要使用验证集,对模型的超参数(神经网络中的神经元数和层数等)进行调整,以及使用正则化的方法,防止模型的过拟合,最终得到训练好的器官芯片药物评价模型。
81.步骤5.2:加载已经训练好的深度学习模型,将测试数据集中的数据作为模型输入,模型的输出即为对器官芯片实验结果的预测。
82.以上仅是本发明众多具体应用范围中的代表性实施例,对本发明的保护范围不构成任何限制。凡采用变换或是等效替换而形成的技术方案,均落在本发明权利保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1