基于深度学习的nanopore测序数据碱基识别方法
1.本发明涉及生物信息学领域,具体为基于深度学习的nanopore测序数据碱基识别方法。
背景技术:
2.oxford公司生产的nanopore第三代测序仪相较于第二代测序仪和pacbio公司的第三代测序仪具有便携、廉价、测序reads长等优点。但是,nanopore的测序准确率远低于二代测序技术和pacbio的hifi测序技术。其官方提供的碱基识别工具准确率只有90%左右,且不开源。nanopore测序仪的纳米孔本质上是一个纳米级的蛋白质孔,两侧有检测电压的装置。工作时使用引物牵引单链的dna/rna穿过纳米孔,不同类型的核苷酸穿过纳米孔时会引起不同的电流变化。测序仪记录所有的电流变化,通过把电信号翻译成对应的碱基序列。nanopore为单分子测序,噪声信号和随机误差对碱基识别的准确率影响较大。nanopore测序仪的下机数据分为fasta和fast5两种。其中fasta是使用官方的碱基识别工具(guppy)进行处理后得到的基因序列,准确率在90%左右。fast5文件包含测序仪获取到的原始电信号文本。以官方工具guppy r9.4为例,单次有5个碱基通过纳米孔,因此存在着45=102种可能基因序列。由于碱基修饰的存在,会出现更加复杂的情况。目前已知的碱基修饰有5mc,如果把5mc作为除a、c、g、t之外第5类的碱基信号,那么单次通过纳米孔的5个碱基就有55=3125种可能的序列。且核苷酸和纳米孔都是纳米级的分子结构,官方碱基识别工具不能很好的通过电信号预测出真实的碱基序列。这是影响nanopore测序准确率的一个主要因素。因此,使用深度学习的相关方法构建模型,对nanopore测序原始数据进行可靠的预测是非常有必要的。
技术实现要素:
3.本发明的目的是:针对现有技术中nanopore测序准确率低的问题,提出基于深度学习的nanopore测序数据碱基识别方法。
4.本发明为了解决上述技术问题采取的技术方案是:
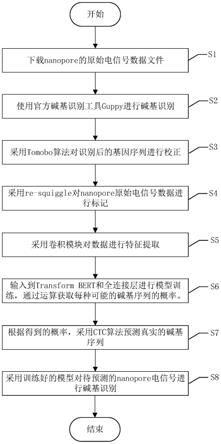
5.基于深度学习的nanopore测序数据碱基识别方法,包括以下步骤:
6.步骤一:下载包括肺杆菌、肠杆菌、变形杆菌在内的50组nanopore原始数据作为训练集;
7.步骤二:对50组原始数据进行碱基识别,得到碱基序列;
8.步骤三:获取准确率在99%以上的illumina测序序列,并以准确率在99%以上的illumina测序序列作为参考基因组,以参考基因组作为ground truth并使用tombo算法对碱基序列进行校正;
9.步骤四:使用re
‑
squiggle方法将校正后的碱基序列转换为对应的电信号数据,然后将电信号数据进行标记;
10.步骤五:利用标记后的电信号数据以及原始数据训练神经网络,并利用训练好的
神经网络进行碱基识别。
11.进一步的,所述神经网络包括第一卷积层、第二卷积层、bert模块、全连接层和ctc解码模块;
12.所述第一卷积层用于对标记后的电信号数据进行降采样,
13.所述第二卷积层用于对降采样后的电信号数据进行特征提取,
14.所述第一卷积层和第二卷积层后设有bn层,所述bn层用于防止均值和方差饱和,
15.所述bert模块用于根据提取到的特征进行训练,并输出电信号数据对应的碱基序列,
16.所述全连接层使用softmax函数对电信号数据对应的碱基序列进行处理,得到原始电信号对应的各个碱基序列的概率,
17.所述ctc解码模块对原始电信号对应的各个碱基序列的概率进行处理,得到最终的碱基序列,
18.所述第一卷积层中卷积核的大小为1
×
3,步长为1
×
2,输出通道为128,
19.所述第二卷积层中卷积核的大小为1
×
3,步长为1
×
2,输出通道为128,
20.所述bert模块包含12层的transformer,768维的embedding隐藏层和12头的注意力机制层。
21.进一步的,所述标记后的电信号数据特征表示为:
[0022][0023]
其中,c表示测序数据,x
c
表示测序数据对应的特征,ω是卷积核的权重,其中参数k设置为3,i和j是序列初始位置,t是序列的长度,x表示累加。
[0024]
进一步的,所述bn层表示为:
[0025][0026]
其中,α、β和∈是模型学到的参数,x
bn
是卷积层输出的序列特征,e是计算期望的函数,var是方差函数。
[0027]
进一步的,所述softmax函数表示为:
[0028][0029]
其中,z
i
表示为第i个节点的输出值,c为分类类别的个数,e表示自然对数函数的底数,为一个数学常数,zc表示第c个节点的输出值。
[0030]
进一步的,所述ctc解码模块具体执行如下步骤:
[0031]
针对bert层输出的预测序列,首先使用beamsearch算法迭代生成候选碱基序列,beam宽度为3,然后对候选碱基进行打分,并去掉碱基序列中的空白字符和冗余字符,选取得分最高的碱基序列作为最终的预测结果,
[0032]
碱基序列存在空白字符的概率为:
[0033][0034]
x是bert层的输出序列,π表示所中间结果对应的路径,β
‑1(l)表示算法搜索过程中所有满足条件的路径,i为输出结果,p(i|x)表示序列中空白字符的概率,
[0035]
利用碱基序列空白字符概率表示ctc损失函数,等于最小化对数域
‑
ln(pπ|x)),ctc损失函数表示为:
[0036][0037]
其中,ln()表示自然对数。
[0038]
进一步的,所述步骤二中对50组原始数据进行碱基识别通过碱基识别工具guppy进行。
[0039]
本发明的有益效果是:
[0040]
(1)本发明使用了性能更好的深度神经网络模型,把解决自然语言处理问题的思路引入到对nanopore测序数据的碱基识别上,与官方的碱基识别工具相比性能更好。
[0041]
(2)本发明为基因组学研究提供了良好的基础,高准确的碱基识别有助于下游基因组数据的分析。
[0042]
(3)本发明的模型泛化性能较好,适用于包括微生物、植物、动物等多物种的nanopore测序数据的碱基识别。
[0043]
本技术利用卷积层对nanopore电信号数据进行下采样和特征提取,使用bert模块预测电信号对应的碱基序列,并使用ctc算法去除冗余数据。实现对nanopore测序数据碱基序列高准确的识别。
附图说明
[0044]
图1为本技术实施例提供的基于深度神经网络模型的nanopore测序数据碱基识别方法流程图;
[0045]
图2为本技术深度神经网络模型的效果示意图;
[0046]
图3为本技术与官方碱基识别工具和guppy
‑
kp在测试集上准确率对比示意图;
[0047]
图4为本技术与官方碱基识别工具在测试集上序列一致性指标对比示意图1;
[0048]
图5为本技术与官方碱基识别工具在测试集上序列一致性指标对比示意图2;
[0049]
图6为本技术与官方碱基识别工具在测试集上序列一致性指标对比示意图3;
[0050]
图7为本技术与官方碱基识别工具在测试集上序列一致性指标对比示意图4;
[0051]
图8为本技术与官方碱基识别工具在测试集上序列一致性指标对比示意图5;
[0052]
图9为本技术与官方碱基识别工具在测试集上序列一致性指标对比示意图6;
[0053]
图10为本技术与官方碱基识别工具在测试集上序列一致性指标对比示意图7;
[0054]
图11为本技术与官方碱基识别工具在测试集上序列一致性指标对比示意图8;
[0055]
图12为本技术与官方碱基识别工具在测试集上序列一致性指标对比示意图9;
[0056]
图13为本技术与官方碱基识别工具在9个物种的测试集上错误率对比示意图。
具体实施方式
[0057]
需要特别说明的是,在不冲突的情况下,本技术公开的各个实施方式之间可以相互组合。
[0058]
具体实施方式一:参照图1具体说明本实施方式,本实施方式所述的基于深度学习的nanopore测序数据碱基识别方法。
[0059]
如图1所示,包括以下步骤s1~s8:
[0060]
s1、下载包括肺杆菌(klebsiella pneumoniae)、肠杆菌(enterobacteriaceae)、变形杆菌(proteobacteria)在内的50组nanopore原始数据和9种真菌的测序数据组成数据集。
[0061]
其中,获取的50组nanopore原始测序数据作为模型的训练集,另外9个物种的基因序列作为测试集。
[0062]
s2、使用nanopore官方的碱基识别工具guppy对50组原始数据进行碱基识别。
[0063]
使用官方碱基识别工具guppy把未知的nanopore转换为碱基序列,用于找到其对应的二代测序的参考基因组。
[0064]
s3、以illumina测序序列作为参考基因组,采用tombo算法对guppy处理后的碱基序列进行校正,并使用动态时间规整算法对校正后的序列进行注释。
[0065]
s4、采用re
‑
squiggle方法把真实的dna序列转换成真实电信号,生成(碱基序列,电信号)格式的标记数据。
[0066]
s5、构建基于卷积神经网络和bert网络的神经网络模型,模型包括两个卷积层、一个bert模块、一个全连接层和一个ctc解码模块。使用卷积模块对输入序列进行特征提取。采用两个卷积层对输入的序列数据进行预处理和特征提取,包括以下步骤:
[0067]
s51、第一个卷积层中卷积核的大小为1
×
3,步长为1
×
2,输出通道为128,用于对数据降采样,减少计算复杂度。
[0068]
s52、第二个卷积层中卷积核的大小为1
×
3,步长为1
×
2,输出通道为128,用于特征提取。输入信号向量x的计算公式如下:
[0069][0070]
s53、每个卷积模块之后都有一个批次标准化(bn)层,用于防止均值和方差饱和,提高模型的泛化性能。计算公式如下:
[0071][0072]
s6、将提取到的特征输入bert模块,经过全连接层处理后输出nanopore原始电信号对应的各个碱基序列的概率。将降采样后提取到的特征输入到bert模块中进行训练。bert模块包含12层的transformer,768维的embedding隐藏层和12头的注意力机制层。紧接着的是一个全连接层,使用softmax函数计算出每个位置上碱基的概率。i和j分别表示序列的序号两个累加符号放在一起xi+j表示序列中的每一个字符卷积之后提取到的序列特征作为后续bert层的输入。计算公式如下:
[0073][0074]
s7、使用ctc解码模块去除重复的碱基序列和空白序列,最终输出高准确率的nanopore碱基序列。使用ctc损失函数计算nanopore原始电信号和碱基序列间的高阶特征分布距离。ctc解码器使用beamsearch算法迭代生成候选碱基序列,beam宽度为3,然后对候选碱基进行打分。在此过程中去掉序列中的空白字符,选取得分最高的碱基序列作为最终的预测结果。通过nanopore的原始信号数据获得高准确率的碱基序列。e是自然对数函数的底数,是一个数学常数。zc表示第c个节点的输出值。碱基序列存在空白字符的概率计算过程和ctc损失函数公式如下所示:
[0075][0076]
l(s)=
‑
lnπ
(x,z)∈s
p(z|x)=
‑
∑
(x,z)∈s
lnp(z|x)
[0077]
其中,x表示输入序列,π表示beamsearch搜索到的碱基的路径,z表示输出序列。
[0078]
s8、采用训练好的预测模型,把nanopore测序的原始电信号转换为准确率高于官方工具的碱基序列。
[0079]
下面以一组具体实验例对本发明的识别效果作进一步描述。
[0080]
首先,为了评价碱基识别工具的性能,我们在相同的数据集上对包括我们的模型在内的4种碱基识别工具进行了对比分析。其中,ourmethod表示该发明的深度神经网络模型,guppy和albacore是oxford公式的官方碱基识别工具,guppy
‑
kp是在官方基础上重新训练后的模型。
[0081]
表一展示了包括我们方法在内的4种工具在测试集上的错误率。
[0082]
其中,deletion、insertion、mismatch分别表示测序数据种的删除错误、插入错误和匹配错误。碱基识别准确率定义如下:
[0083][0084]
m表示匹配到的碱基数量,s表示匹配错误的碱基数,i表示插入错误的碱基数量,d表示删除错误的碱基数。在klebsiella pneumoniae nuh29数据集上,本发明的方法错误率为11.06%,低于其他的碱基识别工具。在klebsiella pneumoniae ksb2数据集上,本发明的方法、albacore、guppy的错误率分别为11.26%、15.80%、15.73%,错误率低于官方提供的碱基识别工具。
[0085]
其次,我们还使用了基因组组装一致性作为评估模型性能的指标。图4展示了包括本发明在内的4种碱基识别工具的共有序列一致性。我们使用了聚合物插入错误、其他插入错误、聚合物删除错误、其他删除错误、替代错误和dcm错误6个指标来评估模型性能。
[0086]
测试集上性能评估表明,本发明的碱基识别错误率和基因组组装一致性指标上都优于官方提供的碱基识别工具。
[0087]
需要注意的是,具体实施方式仅仅是对本发明技术方案的解释和说明,不能以此限定权利保护范围。凡根据本发明权利要求书和说明书所做的仅仅是局部改变的,仍应落入本发明的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1