基于多视图深度学习的化合物-蛋白质相互作用预测方法
基于多视图深度学习的化合物
‑
蛋白质相互作用预测方法
技术领域
1.本发明涉及生物信息处理技术领域,具体涉及一种基于多视图深度学习的化合物
‑
蛋白质相互作用预测方法。
背景技术:
2.阐明化合物
‑
蛋白质相互作用(cpi)的机制在发现和开发安全有效的药物、预测潜在的副作用和为现有药物寻找新的用途,即药物重新定位中起着至关重要的作用。
3.尽管各种实验检测方法已被广泛用于候选药物的筛选和特性描述,但通过实验来识别cpi往往是耗费时间和资源的。采用深度学习的方法预测化合物
‑
蛋白质的相互作用大大降低了药物开发的时间和成本,并保持了高效和高质量的创新药物靶点识别和开发。同时,通过深度学习方法深入分析化合物与蛋白质之间相互作用的内部普遍机制,可更好地提高药物结合的特异性或避免副作用。
4.目前,现有的使用深度学习技术进行cpi预测的方法,如deepcpi、monn、deepaffinity和transformercpi等。例如,zhou等人通过整合化合物的化学指纹和蛋白质的局部特征,开发了dl
‑
cpi,这是一个深度神经网络(dnn)架构,实现了良好的预测性能。zeng及团队提出了一个多模态的深度学习模型即deepcpi,通过潜在语义分析和word2vec特征嵌入技术获得低维表示,并通过多模态dnn预测cpi,取得了良好的预测效果(2019)。后来,zeng等人进一步改进并开发了一个名为monn(a multi
‑
objective neural network for prediculate compound
‑
protein interactions and affinities)的新型模型,采用图卷积网络学习全局和局部特征来预测cpi。同样,masashi等人也构建了一个基于gnn的预测器,称为cpi_prediction,它使用gnn进行转换和神经注意机制来捕捉化合物和蛋白质之间的交互位点。通过引入先进的nlp(自然语言处理)技术,zheng等人开发了transformercpi,这是一个基于transformer的模型,利用修改后的transformer编码器探索transformer结构在cpi预测中的表现,并通过标签反转数据集更接近地模拟真实情况。不同的是,mostafa等人提出了一个名为deepaffinity的半监督模型,使用rnn
‑
cnn网络来利用未标记和标记的数据。
5.尽管上述基于深度学习的方法已经越来越多地被应用于cpi预测,并在一定程度上取得了性能提升的成功,但仍然存在一些需要解决的局限性。首先,现有方法的性能不能满足药物发现的准确预测需求。进一步提高性能是迫切需要做的。其次,现有的方法大多侧重于对蛋白质或化合物的信息提取,而忽略了化合物和蛋白质之间的交互信息的重要性,预测不够精确。
技术实现要素:
6.本发明的目的在于提供一种直接从蛋白质氨基酸序列和化合物smiles(简化分子线性输入规范)的二维结构中预测其相互作用的基于多视图深度学习的化合物
‑
蛋白质相互作用预测方法及系统,以解决上述背景技术中存在的至少一项技术问题。
7.为了实现上述目的,本发明采取了如下技术方案:
8.一方面,本发明提供一种基于多视图深度学习的化合物
‑
蛋白质相互作用预测方法,包括:
9.对于待预测的化合物
‑
蛋白质对,提取其中的蛋白质的序列特征以及化合物的结构特征;
10.将提取的序列特征以及结构特征进行编码融合,获得表征化合物和蛋白质间相互作用的相关性信息的统一特征;
11.利用预先训练好的预测模型,对序列特征、结构特征以及统一特征进行处理,得到蛋白质和化合物是否会发生相互作用的结果;其中,所述预先训练好的预测模型由训练集训练得到,所述训练集包括多个化合物
‑
蛋白质对以及标注每一个化合物
‑
蛋白质对相互作用的标签。
12.优选的,提取蛋白质的序列特征包括:提取原始蛋白质信息,结合多头注意力机制,得到预期维度的特征;基于预期维度的特征,结合滤波函数,计算出蛋白质的序列特征。
13.优选的,提取蛋白质的序列特征包括:使用预训练语言模型bert提取原始蛋白质信息,结合多头注意力机制,得到预期维度的特征;使用卷积神经网络cnn,结合滤波函数,基于预期维度的特征计算出蛋白质的序列特征。
14.优选的,提取化合物的结构特征包括:获取化合物的原子列表、键矩阵和相邻矩阵;基于原子列表、键矩阵和相邻矩阵提取指纹信息;根据指纹信息,得到化合物的结构特征。
15.优选的,提取化合物的结构特征包括:获取化合物的原子列表、键矩阵和相邻矩阵,通过一维wl算法提取指纹信息,将提取的指纹信息作为图卷积网络gnn的输入进行编码,得到化合物的结构特征。
16.优选的,获得统一特征包括:基于自动编码器的多视图学习模型,对蛋白质的序列特征和化合物的结构特征进行编解码,提取编码层和解码层之间的隐藏层的信息,得到统一特征。
17.优选的,结合自动编码器的多视图学习模型的输入
‑
输出相似性比较损失函数和交叉熵损失函数训练所述预测模型。
18.第二方面,本发明提供一种基于多视图深度学习的化合物
‑
蛋白质相互作用预测系统,包括:
19.提取模块,用于对于待预测的化合物
‑
蛋白质对,提取其中的蛋白质的序列特征以及化合物的结构特征;
20.融合模块,用于将提取的序列特征以及结构特征进行编码融合,获得表征化合物和蛋白质间相互作用的相关性信息的统一特征;
21.预测模块,用于利用预先训练好的预测模型,对序列特征、结构特征以及统一特征进行处理,得到蛋白质和化合物是否会发生相互作用的结果;其中,所述预先训练好的预测模型由训练集训练得到,所述训练集包括多个化合物
‑
蛋白质对以及标注每一个化合物
‑
蛋白质对相互作用的标签。
22.第三方面,本发明提供一种非暂态计算机可读存储介质,所述非暂态计算机可读存储介质用于存储计算机指令,所述计算机指令被处理器执行时,实现如上所述的基于多
视图深度学习的化合物
‑
蛋白质相互作用预测方法。
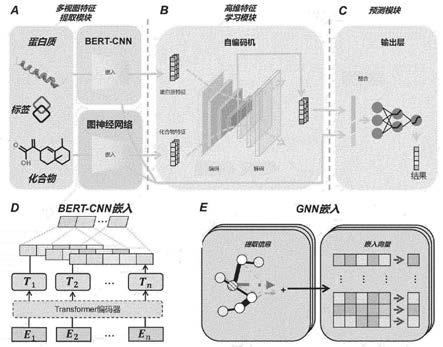
23.第四方面,本发明提供一种电子设备,包括:处理器、存储器以及计算机程序;其中,处理器与存储器连接,计算机程序被存储在存储器中,当电子设备运行时,所述处理器执行所述存储器存储的计算机程序,以使电子设备执行实现如上所述的基于多视图深度学习的化合物
‑
蛋白质相互作用预测方法的指令。
24.本发明有益效果:引入了bert和cnn的新型网络结构进行蛋白质特征嵌入,充分提取了蛋白质和化合物的特征信息,同时,使用gnn来提取化合物的结构特征;引入ae2网络来探索蛋白质和化合物的潜在交互信息,并从不同视角的特征空间生成一个统一的特征空间;将bert
‑
cnn的蛋白质信息、gnn获得的化合物信息和ae2提取的统一信息结合起来,作为下游预测的输入,保持了蛋白质和化合物的原始信息,化合物
‑
蛋白质相互作用预测结果更加准确。
25.本发明附加的方面和优点将在下面的描述中部分给出,这些将从下面的描述中变得明显,或通过本发明的实践了解到。
附图说明
26.为了更清楚地说明本发明实施例的技术方案,下面将对实施例描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
27.图1为本发明实施例中所提出的神经网络的结构,即mdl
‑
cpi的示意图;
28.图2为本发明实施例中bert
‑
cnn的简要结构图;
29.图3为本发明实施例中gnn模块的架构图;
30.图4为本发明实施例中ae2模块的简要结构图;
31.图5为cpi_prediction,transformercpi和本发明实施例中所提出的mdl
‑
cpi三种模型在人类数据集和c.elegans数据集上的pr曲线和roc曲线;
32.图6为cpi_prediction、transformercpi和本发明实施例中提出的mdl
‑
cpi在人类数据集和c.elegans数据集上的整体对比结果;
33.图7为mdl
‑
cpi、mdl
‑
cpi(无ae2)、mdl
‑
cpi(无bert)和mdl
‑
cpi(无bert和ae2)的比较结果;
34.图8为本发明实施例中提出的mdl
‑
cpi(即bert
‑
cnn)、mdl
‑
cpi与bert(即bert)以及mdl
‑
cpi与cnn(即cnn)在人类数据集和c.elegans数据集的pr曲线和roc曲线;
35.图9为本发明实施例中提出的mdl
‑
cpi的pca的特征可视化示意图;
36.图10为不含ae2的mdl
‑
cpi的pca的特征可视化示意图。
具体实施方式
37.下面详细叙述本发明的实施方式,所述实施方式的示例在附图中示出,其中自始至终相同或类似的标号表示相同或类似的元件或具有相同或类似功能的元件。下面通过附图描述的实施方式是示例性的,仅用于解释本发明,而不能解释为对本发明的限制。
38.本技术领域技术人员可以理解,除非另外定义,这里使用的所有术语(包括技术术
语和科学术语)具有与本发明所属领域中的普通技术人员的一般理解相同的意义。
39.还应该理解的是,诸如通用字典中定义的那些术语应该被理解为具有与现有技术的上下文中的意义一致的意义,并且除非像这里一样定义,不会用理想化或过于正式的含义来解释。
40.本技术领域技术人员可以理解,除非特意声明,这里使用的单数形式“一”、“一个”、“所述”和“该”也可包括复数形式。应该进一步理解的是,本发明的说明书中使用的措辞“包括”是指存在所述特征、整数、步骤、操作、元件和/或组件,但是并不排除存在或添加一个或多个其他特征、整数、步骤、操作、元件和/或它们的组。
41.在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。而且,描述的具体特征、结构、材料或者特点可以在任一个或多个实施例或示例中以合适的方式结合。此外,在不相互矛盾的情况下,本领域的技术人员可以将本说明书中描述的不同实施例或示例以及不同实施例或示例的特征进行结合和组合。
42.为便于理解本发明,下面结合附图以具体实施例对本发明作进一步解释说明,且具体实施例并不构成对本发明实施例的限定。
43.本领域技术人员应该理解,附图只是实施例的示意图,附图中的部件并不一定是实施本发明所必须的。
44.实施例1
45.本实施例1提供一种基于多视图深度学习的化合物
‑
蛋白质相互作用预测系统,包括:
46.提取模块,用于对于待预测的化合物
‑
蛋白质对,提取其中的蛋白质的序列特征以及化合物的结构特征;
47.融合模块,用于将提取的序列特征以及结构特征进行编码融合,获得表征化合物和蛋白质间相互作用的相关性信息的统一特征;
48.预测模块,用于利用预先训练好的预测模型,对序列特征、结构特征以及统一特征进行处理,得到蛋白质和化合物是否会发生相互作用的结果;其中,所述预先训练好的预测模型由训练集训练得到,所述训练集包括多个化合物
‑
蛋白质对以及标注每一个化合物
‑
蛋白质对相互作用的标签。
49.本实施例1中,利用上述的基于多视图深度学习的化合物
‑
蛋白质相互作用预测系统,实现了化合物
‑
蛋白质相互作用预测方法,该方法包括:
50.使用提取模块对于待预测的化合物
‑
蛋白质对,提取其中的蛋白质的序列特征以及化合物的结构特征;
51.使用融合模块,将提取的序列特征以及结构特征进行编码融合,获得表征化合物和蛋白质间相互作用的相关性信息的统一特征;
52.使用预测模块,利用预先训练好的预测模型,对序列特征、结构特征以及统一特征进行处理,得到蛋白质和化合物是否会发生相互作用的结果;其中,所述预先训练好的预测模型由训练集训练得到,所述训练集包括多个化合物
‑
蛋白质对以及标注每一个化合物
‑
蛋白质对相互作用的标签。
53.本实施例1中,提取蛋白质的序列特征包括:提取原始蛋白质信息,结合多头注意力机制,得到预期维度的特征;基于预期维度的特征,结合滤波函数,计算出蛋白质的序列特征。具体的,使用预训练语言模型bert提取原始蛋白质信息,结合多头注意力机制,得到预期维度的特征;使用卷积神经网络cnn,结合滤波函数,基于预期维度的特征计算出蛋白质的序列特征。
54.本实施例1中,提取化合物的结构特征包括:获取化合物的原子列表、键矩阵和相邻矩阵;基于原子列表、键矩阵和相邻矩阵提取指纹信息;根据指纹信息,得到化合物的结构特征。具体的,获取化合物的原子列表、键矩阵和相邻矩阵,通过一维wl算法提取指纹信息,将提取的指纹信息作为图卷积网络gnn的输入进行编码,得到化合物的结构特征。
55.本实施例1中,获得统一特征包括:基于自动编码器的多视图学习模型,对蛋白质的序列特征和化合物的结构特征进行编解码,提取编码层和解码层之间的隐藏层的信息,得到统一特征。
56.本实施例1中,结合自动编码器的多视图学习模型的输入
‑
输出相似性比较损失函数和交叉熵损失函数训练所述预测模型。
57.实施例2
58.本实施例2提供一种基于多视图深度学习的化合物
‑
蛋白质相互作用预测系统,包括:
59.提取模块,用于对于待预测的化合物
‑
蛋白质对,提取其中的蛋白质的序列特征以及化合物的结构特征;
60.融合模块,用于将提取的序列特征以及结构特征进行编码融合,获得表征化合物和蛋白质间相互作用的相关性信息的统一特征;
61.预测模块,用于利用预先训练好的预测模型,对序列特征、结构特征以及统一特征进行处理,得到蛋白质和化合物是否会发生相互作用的结果;其中,所述预先训练好的预测模型由训练集训练得到,所述训练集包括多个化合物
‑
蛋白质对以及标注每一个化合物
‑
蛋白质对相互作用的标签。
62.本实施例2中,利用上述的基于多视图深度学习的化合物
‑
蛋白质相互作用预测系统,实现了基于多视角深度学习的化合物
‑
蛋白质相互作用预测方法(mdl
‑
cpi),该方法包括:
63.使用提取模块对于待预测的化合物
‑
蛋白质对,提取其中的蛋白质的序列特征以及化合物的结构特征;
64.使用融合模块,将提取的序列特征以及结构特征进行编码融合,获得表征化合物和蛋白质间相互作用的相关性信息的统一特征;
65.使用预测模块,利用预先训练好的预测模型,对序列特征、结构特征以及统一特征进行处理,得到蛋白质和化合物是否会发生相互作用的结果;其中,所述预先训练好的预测模型由训练集训练得到,所述训练集包括多个化合物
‑
蛋白质对以及标注每一个化合物
‑
蛋白质对相互作用的标签。
66.具体的,如图1所示,本实施例2中的化合物
‑
蛋白质相互作用预测系统,实现了基于多视角深度学习的化合物
‑
蛋白质相互作用预测方法(mdl
‑
cpi),包含三个主要模块:多视图特征提取模块(即提取模块)、高延迟特征学习模块(即融合模块)、预测模块。首先,作
为一个给定的输入化合物和蛋白质对,使用多视图特征提取模块来分别表示蛋白质和化合物特征。利用bert
‑
cnn进行蛋白质序列信息嵌入,利用gnn进行化合物结构信息嵌入。其次,为了充分挖掘化合物和蛋白质之间的交互信息,采用了ae2网络,即一种高延迟的自动编码器技术,从bert
‑
cnn输出的蛋白质特征和gnn输出的化合物特征中生成高度集中的统一特征。最后,在预测模块中,根据所得到的由蛋白质、化合物和统一特征组成的特征,预测相关的蛋白质和化合物是否相互作用。
67.如图2所示,本实施例2中引入了一种多视图特征提取算法,分别用于蛋白质序列信息和化合物结构信息。对蛋白质使用bert和cnn的混合网络(表示为bert
‑
cnn)提取蛋白质序列特征,对化合物利用gnn网络提取化合物结构特征。对于蛋白质信息的提取,首先通过预训练的bert模型将给定的序列编码到嵌入矩阵中(其中和为输入的cpi样本数),初步提取蛋白质序列特征,以避免冗余的序列信息。为了加强特征表示能力,将其送入cnn,从而生成蛋白质特征向量。对于化学结构信息的提取,将化合物信息作为gnn(图神经网络)的输入,然后输出化合物向量。这两个特征向量和可以为下游任务分析提供重要信息。
68.其中,提取蛋白质的序列特征包括:提取原始蛋白质信息,结合多头注意力机制,得到预期维度的特征;基于预期维度的特征,结合滤波函数,计算出蛋白质的序列特征。具体的,使用预训练语言模型bert提取原始蛋白质信息,结合多头注意力机制,得到预期维度的特征;使用卷积神经网络cnn,结合滤波函数,基于预期维度的特征计算出蛋白质的序列特征。
69.对于bert
‑
cnn的嵌入,现有的相关模型的性能证明了bert的力量。给定一个输入的蛋白质序列,可以将一个蛋白质序列分割成一个重叠的3格氨基酸序列。然后,通过编码器层中的多头注意子层和定位前馈网络,将其发送到bert,以生成原始蛋白质信息。
70.为了讨论的方便,向量用粗体小写字母(如),矩阵用粗体大写字母(如),标量则用非粗体字母(如dim=32)。
71.在本实施例的的网络结构中,注意力机制被用来衡量预测句子中每个位置的重要性。它将根据query,key和value(key对应原始句子,query是翻译句子,value是隐藏层向量)来权衡输入和输出的相似性,做隐藏层的加权平均,得到最一致的输出。
[0072][0073]
其中,q、k、v是相同x
word
的,d
k
=dim/n
head
是q或k的嵌入维度,n
head
是多头关注子层的头数的超参数。
[0074]
多头注意机制并行运行多个注意模块,然后将每个独立的注意模块的输出串联起来,通过线性层转换使输出成为预期维度。
[0075][0076]
其中,和是可学习的参数。其中x
multihead
=multihead(x
words
)=(x1,x2,
…
,x
len
)。
[0077]
同时,为了赋予模型以非线性和不同维度之间的相互作用,对自我注意子层的输出,在每个位置分别应用相同的pffn:
[0078]
pffn(x
multihead
)=relu(x
multihead
w
(1)
)w
(2)
[0079]
其中,和是可学习的参数,在所有位置上共享。这些参数在各层之间是不同的。relu(rectified linear unit)是一个非线性激活函数。
[0080]
综上所述,对每个bert编码器层的计算过程如下。
[0081][0082]
其中,t代表第t个bert编码器层;是x
words
输入。
[0083]
最后一个bert编码器层的输出中的是作为整个bert的输出。此外,本实施例中还采用了dropout技术来防止bert的过拟合。
[0084]
为了进一步提取蛋白质特征信息,本实施例2引入了cnn,它可以获得bert输出的低维表示。使用滤波函数,cnn从bert输出和权重矩阵(学习参数)中计算出一个隐藏向量,然后产生输出向量。
[0085]
本实施例的cnn使用一个滤波函数,其中输入是输出是一个隐藏向量,表示如下:
[0086][0087]
其中,w
conv
是权重矩阵,b
conv
是偏置向量。请注意,这个滤波函数允许从一个t维的输入向量获得一个t维的隐藏向量。因此,可以分层次地应用这个函数,即我们计算第t个隐藏向量为来得到一组隐藏向量:其中|c|是向量的数量。为了从这组隐藏向量中获得最终输出使用以下的平均值:
[0088][0089]
本实施例2中,提取化合物的结构特征包括:获取化合物的原子列表、键矩阵和相邻矩阵;基于原子列表、键矩阵和相邻矩阵提取指纹信息;根据指纹信息,得到化合物的结构特征。如图3所示,具体的,获取化合物的原子列表、键矩阵和相邻矩阵,通过一维wl算法提取指纹信息,将提取的指纹信息作为图卷积网络gnn的输入进行编码,得到化合物的结构特征。
[0090]
具体的,对于化合物的输入,本实施例2中首先通过rdcit获得了长度为(化合物中的节点数)的原子列表dict
atom
、尺寸为n
atom
×
n
atom
的键矩阵w
bond
和尺寸为n
atom
×
n
atom
的相邻矩阵w
adjancency
。然后,通过一维weisfeiler
‑
lehman算法(1
‑
wl)的思路提取其指纹信息,为
gnn特征提取过程提供输入。之后,通过使用单次嵌入,指纹被编码为一个大小为n
atom
×
dim的矩阵x
fingerprints
。接下来,被送入gnns以提取复合结构特征,即:
[0091][0092]
其中,t代表第t层gnn;relu(rectified linear unit)是一个非线性激活函数;w
gnn
是权重矩阵,大小为dim
×
dim。
[0093]
为了得到gnn最后一层的输出,使用了如下的每一列
[0094]
的平均值。
[0095][0096]
其中,x
i
表示gnn最后一层中化合物的第i个节点的向量表现;n
atom
是化合物中的节点数。
[0097]
本实施例2中,获得统一特征包括:基于自动编码器的多视图学习模型,对蛋白质的序列特征和化合物的结构特征进行编解码,提取编码层和解码层之间的隐藏层的信息,得到统一特征。
[0098]
具体的,如图4所示,ae2是一个基于自动编码器的多视图学习模型,自动编码器是一个在编码器和解码器之间有一个内部隐藏层的神经网络,以无监督的方式训练。本实施例的ae2在编码和解码后提高输入和输出的相似度,然后提取隐藏层的信息作为输出。通过编码和解码的过程,隐藏层的低维高集中数据必须包含编码网络所获得的输入数据的主要cpi相关特征。请注意,中间隐藏层的高集中数据是我们在cpi预测中需要的统一特征信息。
[0099]
受ae2的启发,定义多视图数据x={x
(1)
,x
(2)
},其中和是各视图的特征呈现。对第v个视图的ae2网络表示为ae
‑
net(内部ae网络),用于每个视图的特征,dg
‑
net(退化网络)用于将完整的潜在表征映射回每个视图。
[0100]
关于ae
‑
net,第一个m/2(其中是m是线性层的数量)隐藏层将输入编码为新的表征,最后几层对表征进行解码以重建输入。给定一个输入特征向量z
(0,v)
=x
(v)
,其中v表示第v个视图,那么第m层(m=1,2,
…
,m)的输出为
[0101][0102]
其中,和d(m,v)是第v层对第v个视图的节点数。和分别表示与第m层相关的权重和偏置,是用于调整的参数。然后,给定第v个视图的特征向量x
(v)
,相应的重构表征被表示为z
(m,v)
,得到的低维表征是潜在表征h。关于dg网,更新潜在表示h,类似地,第,层(l=1,2,
…
,m)的输出为和g
(0,v)
=h。
[0103]
蛋白质特征和复合特征分别通过ae网和dg网,并进行两次前向传播和更新,最后
整个ae2的输出v
i_unified
为h,网络的目标为:
[0104][0105]
本实施例2中,结合自动编码器的多视图学习模型的输入
‑
输出相似性比较损失函数和交叉熵损失函数训练所述预测模型。
[0106]
具体的,为了充分利用统一特征,尽量保留蛋白质和化合物的特征,将蛋白质特征、化合物特征和统一特征信息结合起来,并将合并后的特征送入预测模块,预测相关蛋白质和化合物是否会发生相互作用。
[0107]
y=w
output
[v
i_compound
+v
i_protein
+v
i_unified
]+b
output
[0108]
其中w
output
、b
output
分别表示权重和偏差,v
i_protein
是蛋白质特征(来自bert
‑
cnn),v
i_compound
·
是化合物特征(来自gnn),v
i_unified
是统一的融合特征(来自ae2),其中,i∈1,2,...,n和n是输入cpi样本的数量。
[0109]
为了训练一个稳健的预测模型并提高其性能,结合两个成本损失函数:ae2的输入
‑
输出相似性比较损失函数和预测模块的交叉熵损失函数,如下所示。
[0110][0111]
其中,第一个公式是ae2的损失函数,如上所述。第二式是交叉熵损失函数,x[j]是预测样本的类别,x是真实类别。
[0112]
在实施例2中,用以下五个常用的指标评估了本实施例2的方法和其他现有模型的性能。acc(准确率)、精确率、召回率、prc(精确率
‑
召回率曲线下的面积)、auc(接受者操作特征曲线下的面积)。
[0113]
它们的计算公式如下:
[0114][0115]
其中tp、fn、tn和fp分别代表真阳性、假阴性、真阴性和假阳性样本的数量。acc是一个综合指标,通常描述模型在所有类别中的表现,在cpi预测中非常有用。精确率指的是预测中正确的例子数与阳性类总数的比例,它反映了模型在将样本分类为阳性时的可靠程度。召回率指的是正确分类为阳性的样本数与阳性样本总数的比例,它反映了模型在检测
阳性样本的能力上是否可以信赖。roc(receiver operating characteristic)曲线下的面积被称为auc(area under roc curve),它被用来衡量模型的性能。同样,pr(精确率和召回率)曲线下的面积被称为prc(pr曲线下面积)。一般来说,acc、precision、recall、auc和prc都在0.5和1之间,这些指标越高,说明模型的分辨能力越强,预测效果越好。
[0116]
严格的数据集对于训练有效的、有前途的预测器是根本性的关键。在基于机器学习的cpi预测研究中,用于模型训练的数据集通常包含阳性样本(即实验验证的化合物
‑
蛋白相互作用)和阴性样本(验证的非化合物
‑
蛋白相互作用)。值得注意的是,在大多数现有的研究中,生成阴性样本的常用方法是随机洗出真实的cpi。然而,随机阴性样本有可能包括未知的阳性样本,这将使模型的性能在不同的数据集上有很大的不同,也会对实际预测产生负面影响。因此,在数据构建中选择真实可信的负样本是非常重要的。
[0117]
本实施例2中,选择了两个cpi数据集(即人类和c.elegans),这两个数据集最初是由liu等人提出的,因为它们在药物研究和开发中具有普遍性和有效性。他们对阴性样本中每个化合物(或蛋白质)对应的蛋白质(或化合物)进行统计测试。因此,其数据集包括高度可信的化合物
‑
蛋白质对的阴性样本。总之,使用了平衡的cpi数据集,其正负样本的比例(正:负)约为1:1,如表1所示。
[0118]
表1
[0119][0120]
本实施例2中的预测方法与现有的其他方法的比较实验结果:
[0121]
为了评估本实施例dml
‑
cpi的有效性,首先在人类数据集和c.elegans数据集上与一些传统的机器学习方法进行比较,如k
‑
nn(k
‑
nearest neighbor)、随机森林(rf)(breiman,2001)、二级logistic(l2)和svm等。这些方法使用基于pubchem指纹和pfam域的特征,分别使用k
‑
nn、rf、l2和svm作为分类器。表2、表3分别说明了人类数据集和c.elegans数据集的比较结果。"
‑
"代表没有参考数据。在表2、表3中,最佳结果以粗体字显示。
[0122]
表2
[0123][0124]
如表2所示,可以看到,mdl
‑
cpi在人类数据集上取得了比机器学习方法更好的性
能。具体来说,mdl
‑
cpi实现了96.1%的auc,与rf相比,产生了2.1%的相对改善。
[0125]
表3
[0126][0127]
同样地,如表3所示,mdl
‑
cpi在大多数情况下取得了更高的性能。具体来说,mdl
‑
cpi的auc为97.0%,精确度为94.7%,召回率为88.2%,与rf相比有7.0%的相对改善,与l2相比有5.3%,与l2相比有4.2%,这表明mdl
‑
cpi与这些基于机器学习的方法相比具有优势。mdl
‑
cpi和这些基于机器学习的方法的比较结果证明了所提模型的优越性。由于通过bert
‑
cnn全面提取蛋白质特征,通过gnn有效提取化合物特征,通过多视图ae2生成关联信息,mdl
‑
cpi使用的特征更具有鉴别性和代表性。同时,本实施例是数据驱动的,不需要像svm那样使用化学先验知识。因此,mdl
‑
cpi在预测cpi方面比这些模型有更好的效果。
[0128]
接下来,通过比较本实施例mdl
‑
cpi与其他现有的基于深度学习的方法,即cpi_prediction和transformercpi,来研究本实施例dml
‑
cpi的有效性。从表2和表3可以看出,mdl
‑
cpi在所有评估指标方面都取得了优异的成绩,只有一个例外。在人类数据集上,本实施例给出了最佳的auc为96.1%,acc为91.0%,精确率为92.4%,召回率为90.5%,与cpi_prediction相比,分别产生了1.4%、1.5%、1.7%和0.8%的相对改进。同样,在c.elegans数据集上,提议的mdl
‑
cpi的auc、acc和精度比cpi_prediction高0.5、0.6和1.0%。由此可见,mdl
‑
cpi在不同物种数据集上分别优于其他方法。提出的mdl
‑
cpi使用bert
‑
cnn提取的代表性蛋白质特征和gnn映射的有效化合物特征,并加入ae2来捕捉蛋白质特征和化合物特征的关联信息,这就避免了蛋白质和化合物之间重要的交互信息的损失。因此,与其他现有模型相比,mdl
‑
cpi取得最佳性能并不令人惊讶。
[0129]
为了直观地比较本发明的实施例和其他模型,进一步比较了拟议的mdl
‑
cpi和其他基于深度学习的方法的roc和pr曲线,如图5所示,可以看到,dml
‑
cpi具有最高的prc和auc,在人类数据集上分别为0.962、0.959,在c.elegans数据集上为0.973、0.975。这表明本实施例的模型在预测cpi方面有很强的性能。
[0130]
为了进一步比较本发明的实施例与其他模型的整体预测情况,用对比图来清楚地显示本实施例在各个评价标准下的优异表现。如图6所示,本实施例的预测结果相对集中在一个较高的水平上,本实施例具有较强的抗噪声性能,优于其他两个模型。
[0131]
本实施例2中,研究了bert和cnn对性能的影响:
[0132]
为了直观地证明bert
‑
cnn在本实施例中的优秀嵌入性能,进一步比较了所提出的mdl
‑
cpi(含bert
‑
cnn)与相关变形模型的roc和pr曲线,如(1)含bert的mdl
‑
cpi,与原始mdl
‑
cpi类似,但只使用bert提取蛋白质特征(devlin,等,2018);(2)含cnn的mdl
‑
cpi,同样地,只使用单热编码和cnn来提取蛋白质特征。如图7所示,其中,图7(a)中,横坐标由左向右
依次为auc、acc、prc,在auc、acc、prc中对应的纵坐标的灰色条,由左向右依次为mdl
‑
cpi、mdl
‑
cpi(without ae2)、mdl
‑
cpi(without bert)、mdl
‑
cpi(without bert and ae2);图7(b)中,横坐标由左向右依次为auc、acc、prc,在auc、acc、prc中对应的纵坐标的灰色条,由左向右依次为mdl
‑
cpi、mdl
‑
cpi(without ae2)、mdl
‑
cpi(without bert)、mdl
‑
cpi(without bert and ae2)。由图7可以看出,与使用cnn的dml
‑
cpi相比,使用bert
‑
cnn的dml
‑
cpi具有最高的prc和auc,在人类数据集上产生了2.1%和0.8%的相对改善,在c.elegans数据集上产生了0.4%和1.4%的相对改善。这证明了bert
‑
cnn在提取蛋白质特征方面的强大性能。
[0133]
为了说明本实施例mdl
‑
cpi如何学习有效的特征表示,使用pca和t
‑
sne图来可视化特征表示,以进一步研究特征空间从初始到稳定状态的分布变化,用颜色标记来区分化合物
‑
蛋白质相互作用或不相互作用。图8显示了人类数据集和c.elegans数据集上阳性和阴性样本在二维特征空间的pca分布。图8(a),(c)显示了初始特征空间,而图8(b),(d)对应于稳定的特征空间。可以观察到,在初始特征空间中,正负样本分布在两个没有明显边界的聚类中,用原始特征表示很难直观地区分每个样本。通过mdl
‑
cpi特征迭代过程,特征空间接近于相对稳定,其中特征空间中正负之间的边际分离比较明显。这表明,从mdl
‑
cpi中学习到的特征能更好地区分cpi中的正样本(交互)和负样本(非交互)。
[0134]
此外,为了进一步比较来自ae2的统一特征的必要性,将本实施例与没有ae2的mdl
‑
cpi进行了比较,即mdl
‑
cpi只使用gnn和bert
‑
cnn提取的特征进行预测,而不使用ae2的统一特征。图9展示了本实施例mdl
‑
cpi在历时[0]和[100]的视觉特征空间分布,而图10展示了没有ae2的mdl
‑
cpi在历时[0]和[100]的视觉特征空间分布。这些比较结果表明,由ae2获得的统一特征对性能的提高是有用的。在整体特征中发现的负面和正面样本与ae2的统一特征相结合,可以很容易地区分出来,从而丰富了性能。
[0135]
实施例3
[0136]
本发明实施例3提供一种非暂态计算机可读存储介质,所述非暂态计算机可读存储介质用于存储计算机指令,所述计算机指令被处理器执行时,实现如上所述的基于多视图深度学习的化合物
‑
蛋白质相互作用预测方法,该方法包括:
[0137]
对于待预测的化合物
‑
蛋白质对,提取其中的蛋白质的序列特征以及化合物的结构特征;
[0138]
将提取的序列特征以及结构特征进行编码融合,获得表征化合物和蛋白质间相互作用的相关性信息的统一特征;
[0139]
利用预先训练好的预测模型,对序列特征、结构特征以及统一特征进行处理,得到蛋白质和化合物是否会发生相互作用的结果;其中,所述预先训练好的预测模型由训练集训练得到,所述训练集包括多个化合物
‑
蛋白质对以及标注每一个化合物
‑
蛋白质对相互作用的标签。
[0140]
实施例4
[0141]
本发明实施例4提供一种计算机程序(产品),包括计算机程序,所述计算机程序当在一个或多个处理器上运行时,用于实现如上所述的基于多视图深度学习的化合物
‑
蛋白质相互作用预测方法,该方法包括:
[0142]
对于待预测的化合物
‑
蛋白质对,提取其中的蛋白质的序列特征以及化合物的结
构特征;
[0143]
将提取的序列特征以及结构特征进行编码融合,获得表征化合物和蛋白质间相互作用的相关性信息的统一特征;
[0144]
利用预先训练好的预测模型,对序列特征、结构特征以及统一特征进行处理,得到蛋白质和化合物是否会发生相互作用的结果;其中,所述预先训练好的预测模型由训练集训练得到,所述训练集包括多个化合物
‑
蛋白质对以及标注每一个化合物
‑
蛋白质对相互作用的标签。
[0145]
实施例5
[0146]
本发明实施例5提供一种电子设备,包括:处理器、存储器以及计算机程序;其中,处理器与存储器连接,计算机程序被存储在存储器中,当电子设备运行时,所述处理器执行所述存储器存储的计算机程序,以使电子设备执行实现如上所述的基于多视图深度学习的化合物
‑
蛋白质相互作用预测方法的指令,该方法包括:
[0147]
对于待预测的化合物
‑
蛋白质对,提取其中的蛋白质的序列特征以及化合物的结构特征;
[0148]
将提取的序列特征以及结构特征进行编码融合,获得表征化合物和蛋白质间相互作用的相关性信息的统一特征;
[0149]
利用预先训练好的预测模型,对序列特征、结构特征以及统一特征进行处理,得到蛋白质和化合物是否会发生相互作用的结果;其中,所述预先训练好的预测模型由训练集训练得到,所述训练集包括多个化合物
‑
蛋白质对以及标注每一个化合物
‑
蛋白质对相互作用的标签。
[0150]
本领域内的技术人员应明白,本发明的实施例可提供为方法、系统、或计算机程序产品。因此,本发明可采用完全硬件实施例、完全软件实施例、或结合软件和硬件方面的实施例的形式。而且,本发明可采用在一个或多个其中包含有计算机可用程序代码的计算机可用存储介质(包括但不限于磁盘存储器、cd
‑
rom、光学存储器等)上实施的计算机程序产品的形式。
[0151]
本发明是参照根据本发明实施例的方法、设备(系统)、和计算机程序产品的流程图和/或方框图来描述的。应理解可由计算机程序指令实现流程图和/或方框图中的每一流程和/或方框、以及流程图和/或方框图中的流程和/或方框的结合。可提供这些计算机程序指令到通用计算机、专用计算机、嵌入式处理机或其他可编程数据处理设备的处理器以产生一个机器,使得通过计算机或其他可编程数据处理设备的处理器执行的指令产生用于实现在流程图一个流程或多个流程和/或方框图一个方框或多个方框中指定的功能的装置。
[0152]
这些计算机程序指令也可存储在能引导计算机或其他可编程数据处理设备以特定方式工作的计算机可读存储器中,使得存储在该计算机可读存储器中的指令产生包括指令装置的制造品,该指令装置实现在流程图一个流程或多个流程和/或方框图一个方框或多个方框中指定的功能。
[0153]
这些计算机程序指令也可装载到计算机或其他可编程数据处理设备上,在计算机或其他可编程设备上执行一系列操作步骤以产生计算机实现的处理,从而在计算机或其他可编程设备上执行的指令提供用于实现在流程图一个流程或多个流程和/或方框图一个方框或多个方框中指定的功能的步骤。
[0154]
上述虽然结合附图对本发明的具体实施方式进行了描述,但并非对本发明保护范围的限制,所属领域技术人员应该明白,在本发明公开的技术方案的基础上,本领域技术人员在不需要付出创造性劳动即可做出的各种修改或变形,都应涵盖在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1