一种基于单细胞转录组数据的转录因子调控网络建模方法
本发明属于生物技术行业领域,涉及一种利用单细胞组学数据解析多维分子协同调控机制的方法,具体地说,是一种利用多表型单细胞组学数据,基于多项非负矩阵因式分解算法框架,识别表型变化相关的转录因子-基因功能协同调控互作机制的新方法。
背景技术:
1、近年来,受益于微流体芯片技术的发展,单细胞水平的组学测序技术在越来越多基础实验室和临床研究中得到推广应用。其中单细胞转录组测序技术(scrna-seq)因能通过转录表达谱反映单个细胞的即时生理状态,最为受到生物医学领域研究者的关注。随着scrna-seq技术测序深度和精度的不断优化以及一次性可测细胞数目的不断增多,scrna-seq不仅被用于不同细胞类型、细胞亚群的鉴定,细胞不同状态的区分,还被用于细胞谱系的追踪以及细胞在发育、分化等转化过程中关键特征的捕捉。虽然scrna-seq公共数据和工具都在井喷式增长,但目前只有零星研究对scrna-seq数据中的多维分子协同调控信息进行整合提取,尚无基于单细胞组学数据提取表型相关转录因子-基因功能协同调控模块的方法及工具。
2、转录因子是生物体内调控细胞命运、发育模式和特定生物学功能的一类dna结合蛋白,能通过特异性识别真核基因顺式作用元件来调控下游靶基因的转录或激活,进而促进生物表型或生理状态的变化。很多转录因子,如p53,creb等与重要疾病如肿瘤、炎症的发生和发展过程密切相关。了解特定表型变化下转录因子介导的信号途径变化和靶基因表达变化是解析变化背后分子作用机制的重要一环。现存多表型单细胞转录组分析往往只针对同类细胞群体在不同表型下的转录组进行差异基因分析,并直接在该结果基础上开展下游功能分析。但单细胞组学数据通常具有数据稀疏和高噪音的特点,只依靠比较组学手段获得的表型相关分子信息,无法直观反映表型变化相关的调控子活性变化,也无法完整反映表型变化相关的转录因子介导的信号通路或基因功能发生的改变。
3、为研究表型相关的转录因子-基因功能之间的协同调控作用,本发明建立了一种基于多表型单细胞数据识别多维分子协同调控机制的新方法。本方法引入了一项多项非负矩阵因式分解技术。该技术是非负矩阵因式分解技术(non-negative matrixfactorization,nmf)的一类衍生。它依赖先验的分子间互作关系网络以利用已有生物学知识,通过对单细胞数据高维特征的降维提取,实现从单细胞数据中提取与表型变化相关的转录因子-基因功能协同互作信息。和常见非负矩阵分解技术相比,多项非负矩阵因式分解技术依赖先验的分子互作关系,因此基于该技术设计的多维分子协同调控机制识别方法对单细胞数据噪音有更强抗扰性,所识别的多维分子协同调控机制有更好的生物鲁棒性。
技术实现思路
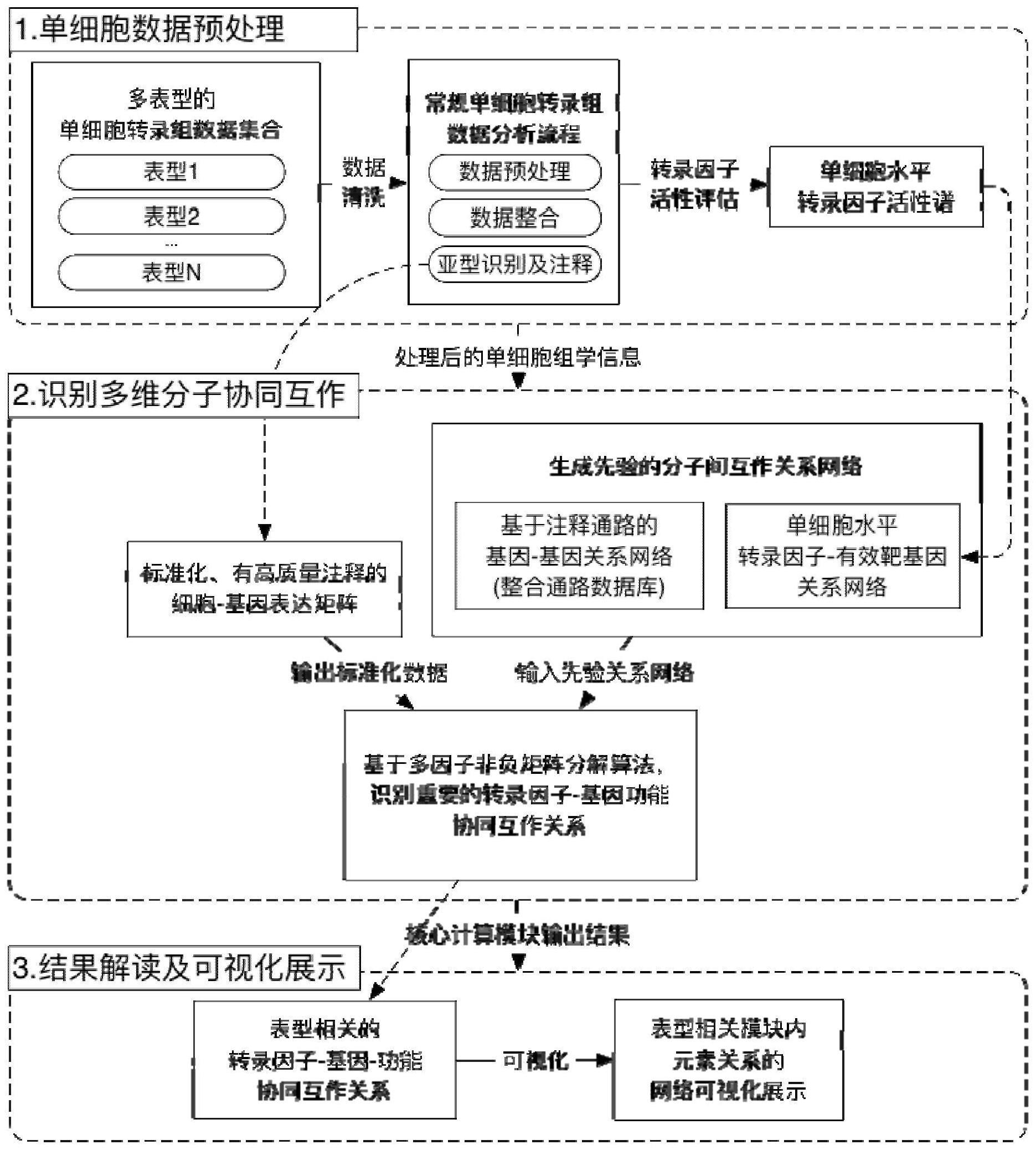
1、本发明的目的在于借助单细胞测序技术和已有的先验生物学知识,提取高维单细胞组学数据内的多维分子协同调控关系,获得与表型变化密切相关的转录因子-基因功能协同调控互作机制,并以一种易读和整合紧密的形式呈现与表型变化相关的多维分子互作关系。
2、本发明公开了一种基于单细胞转录组数据的转录因子调控网络建模方法,包括步骤如下:
3、s1.提取多表型单细胞组学数据,进行数据清洗,对清洗后的数据进行数据整合;
4、s2.基于生物学知识库,对s1处理后的数据进行分析,构建先验的分子间互作关系网络;
5、s3.基于多因子非负矩阵分解算法,根据s1处理后的数据和s2先验的分子间互作关系网络建立多维分子协同互作关系模块;
6、s4.计算多维分子协同互作关系模块与表型相关的互作关系模块;
7、s5.对多维分子协同互作关系模块与表型相关的互作关系模块进行可视化导出。
8、进一步地,所述s1中数据清洗包括:
9、s101设定过滤条件;所述过滤条件至少包括多表型单细胞组学数据中低丰富度的细胞、被细胞碎片污染的细胞、凋亡或裂解的细胞、多聚体中的一种;
10、s102根据过滤条件对多表型单细胞组学数据进行过滤,获得过滤后的数据;
11、s103对过滤后的数据进行特征识别,将识别后的数据进行数据整合;所述特征识别至少包括细胞分群、细胞特征基因识别。
12、进一步地,所述s2中分子间互作关系网络包括转录因子与靶基因之间的调控关系网络、基因与基因之间的功能关联网络。
13、更进一步地,所述基因与基因之间的功能关联网络构建中关联基因至少参与包括调控同一生物学过程、参与同一基因通路、对同一表型有响应中的一种;
14、所述基因与基因之间的功能关联网络中功能关联形式包括但不限于共表观遗传修饰的基因关联网络、基因编码蛋白的配受体互作关系网络、基因编码蛋白的蛋白质相互作用网络中的一种或多种。
15、更进一步地,所述多因子非负矩阵分解算法为:
16、s301设定观测到的总细胞数为n、观测到的总基因个数为m、观测到的总转录因子个数为s,建立一个n×m维的非负矩阵,设定为多表型的单细胞基因表达谱数据x1;建立一个n×s维的非负矩阵,设定为单细胞调控子活性矩阵数据x2;
17、s302设定n个细胞中观察到的所有转录因子-基因功能协同互作模块个数为k,建立一个n×k维的非负矩阵w;建立一个k×m维非负矩阵,用于描述低维空间中变量与基因之间的权重关系,设为h1;建立一个k×s维非负矩阵,用于描述低维空间中变量与转录因子之间的权重关系,设为h2;
18、w满足xi≈whi,hi≥0,w≥0,i=1,2;且分解的平方误为:
19、
20、其中‖■‖f为frobenius范数,基于平方误,构建目标函数:
21、
22、更进一步地,所述s302中式可进一步写为:
23、
24、其中λi为迹系数,tr(hiahjt)和tr(hibhjt)为矩阵的迹矩阵,a为先验基因-基因功能关系的邻接矩阵,b为先验转录因子-靶基因功能关联的邻接矩阵,hj为h1矩阵中第i列向量,hj′为h2矩阵中第j列向量;
25、先验关系邻接矩阵的最大化目标函数为:
26、
27、o2=∑ijbij(hi′2)thj2=tr(h2bh1t)。
28、进一步地,所述s4中计算多维分子协同互作关系模块与表型相关的互作关系具体为:采用差异显著性统计检验方法,检测多因子非负矩阵因式分解后获得低维空间上反映多维分子协同互作关系的变量与表型变化之间的相关性,获得多维分子协同互作关系模块与表型相关的互作关系模块。
29、更进一步地,所述差异显著性统计检验方法包括但不限于学生t检验、mann-whitney u检验、方差分析中的一种。
30、进一步地,所述s5中可视化导出具体为:使用s4所获得多维分子协同互作关系模块与表型相关的互作关系模块,根据对应的转录因子系数矩阵和基因系数矩阵,配合s2中先验的分子间互作关系网络,生成可视化转录因子-基因功能协同关系网络。
31、更进一步地,所述可视化转录因子-基因功能协同关系网络包括若干个协同关系网络节点,所述协同关系网络节点由多个转录因子、多个基因、多个生物功能组成,所述可视化转录因子-基因功能协同关系网络的边缘由转录因子-基因调控关系、基因-基因互作关系和基因-基因功能关系组成。
32、本发明与现有技术相比,基于先验生物学知识对单细胞组学数据的多维分子互作信息进行提取,获得的低维空间上的多维分子互作信息对单细胞组学数据噪音的抗扰性强、生物鲁棒性好;应用可拓展性强,可移植性好,可根据研究背景的不同,灵活调整分子间先验的互作关系网络,提取单细胞数据中与研究目的相关的多维分子调控机制,为解释表型变化背后潜在生物学分子作用机制及表型干预提供手段及工具。
- 还没有人留言评论。精彩留言会获得点赞!