注释宏病毒组原始测序数据短读序列的方法、系统、存储介质和装置
1.本发明涉及生物信息学领域,尤其涉及注释宏病毒组原始测序数据短读序列的方法、系统、存储介质和装置。
背景技术:
2.被称为人体第二基因组的人体微生物组((human microbiome)是人体内外表面所有微生物所携带遗传物质的总称(sender r,et al,2016)。全球各国对这一新兴领域异常重视,并启动了多项微生物组研究计划,例如,由法国农业研究院2005牵头发起并于2008年正式成立了国际人体微生物组联盟;2008年,由美国国立卫生研究院主导启动了人体微生物组计划(human microbiome project,hmp)(meth
éꢀ
barbara a,et al,2012);及欧盟于2008年启动的“人体肠道微生物宏基因组学(metagenomics of human intestinal tract metahit)计划”,在其第七框架计划下,欧盟将目光聚焦在了特定的微生物组领域。这些计划的开展,揭示了人体微生物复杂的组成成分(图1),包括细菌、真菌、人感染病毒、专一性感染细菌的噬菌体等,这些微生物之间相互作用,维持人体内环境的动态平衡,影响着人体的健康。
3.宏病毒组学(viral metagenomics)是宏基因组学的一个分支,但与传统的宏基因组学概念不同,它是在宏基因组学概念的基础上,结合病毒自身的特点,将宏基因组学方法应用到病毒学领域而形成的。2002年,breitbart等(breitbart m,et al,2002)将宏基因组学方法应用于海洋病毒群落的研究,发现噬菌体为海水中主要病毒组,这一研究标志着宏病毒组学正式应用于科学研究。其中,病毒宏基因组测序又称宏病毒组(virome),是在宏基因组学理论的基础上,结合现有的病毒分子生物学检测技术而兴起的一个新的学科分支。宏病毒组直接以环境中所有病毒的遗传物质为研究对象,能够快速准确的鉴定出环境中所有的病毒组成,在病毒发现、病毒溯源、微生物预警等研究方面具有重要作用。宏病毒研究可应用于人或动物肠道或者血液样本、海洋、土壤等的研究,用以挖掘潜在的对人类和环境的危害。
4.宏病毒组测序原始数据是一种压缩的文本文件,其内容包含被测文库的核酸序列以及测序质量的分值,需要进一步将核酸序列注释到数据库中以进行病毒种的鉴定以及后续定量分析。
5.由于很多微生物研究人员或者临床工作者不具备生物信息学知识,很多基层实验室不具备服务器,像metawrap、drvm、virmap等大多序列比对程序需要在linux平台上运行,且需要较高的计算资源,即目前宏病毒组测序数据的注释和提取无法在windows平台上实现,科研人员可能会花费数周时间等待第三方公司出具物种鉴定报告,具有周期长、费用高的弊端,无法做到“利用高通量测序快速鉴定微生物种”的能力,极大阻碍科研进展,拖慢临床检测速度。
技术实现要素:
6.本发明的目的在于克服现有技术的不足,提供一种注释宏病毒组原始测序数据短读序列的方法、系统、存储介质和装置。
7.本发明的目的是通过以下技术方案来实现的:
8.本发明的第一方面,提供一种注释宏病毒组原始测序数据短读序列的方法,基于windows系统,包括以下步骤:
9.获取原始测序数据,对原始测序数据进行解压并提取短读序列,生成短读序列数据集,调用blastn程序将数据集比对到数据库中;
10.保留每条短读序列的最佳比对结果,并将最佳比对结果中比对质量差的结果去除,然后按照基因id添加病毒种名称;
11.基于脚本库,统计注释到每个病毒种的短读序列数目,计算每个病毒种的短读序列在基因组上比对位置的标准偏差,并按照注释的病毒种提取对应的短读序列序列,输出fasta数据集。
12.进一步地,所述方法还包括以下初始化步骤:
13.检测特定目录下是否存在判断是否存在blastn索引,如果不存在blastn索引则进一步判断特定目录下是否存在用户提供的fasta数据集,如果存在用户提供的fasta数据集则基于所述fasta数据集构建新的blastn索引,否则进一步判断特定目录下是否存在refseq病毒数据库,如果存在refseq病毒数据库则基于所述refseq病毒数据库构建新的blastn索引,否则报错;在存在blastn索引的情况下,完成后续注释步骤。
14.进一步地,所述脚本库包括数据库自检脚本、毒株名添加脚本和短读序列提取脚本。
15.进一步地,所述质量差为比对长度小于第一长度和/或相似度小于第一阈值。
16.本发明的第二方面,提供一种注释宏病毒组原始测序数据短读序列的系统,基于windows系统,包括:
17.blastn分析模块:用于获取原始测序数据,对原始测序数据进行解压并提取短读序列,生成短读序列数据集,调用blastn程序将数据集比对到数据库中;保留每条短读序列的最佳比对结果,并将最佳比对结果中比对质量差的结果去除,然后按照基因id添加病毒种名称;
18.短读序列提取及统计分析模块:用于基于脚本库,统计注释到每个病毒种的短读序列数目,计算每个病毒种的短读序列在基因组上比对位置的标准偏差,并按照注释的病毒种提取对应的短读序列序列,输出fasta数据集。
19.进一步地,所述系统还包括:
20.数据库更新模块,用于检测特定目录下是否存在判断是否存在blastn索引,如果不存在blastn索引则进一步判断特定目录下是否存在用户提供的fasta数据集,如果存在用户提供的fasta数据集则基于所述fasta数据集构建新的blastn索引,否则进一步判断特定目录下是否存在refseq病毒数据库,如果存在refseq病毒数据库则基于所述refseq病毒数据库构建新的blastn索引,否则报错;在存在blastn索引的情况下,完成后续注释步骤。
21.进一步地,所述脚本库包括数据库自检脚本、毒株名添加脚本和短读序列提取脚本。
22.进一步地,所述质量差为比对长度小于第一长度和/或相似度小于第一阈值。
23.本发明的第三方面,提供一种存储介质,其上存储有计算机指令,所述计算机指令运行时执行所述的一种注释宏病毒组原始测序数据短读序列的方法的步骤。
24.本发明的第四方面,提供一种装置,包括存储器和处理器,所述存储器上存储有可在所述处理器上运行的计算机指令,所述处理器运行所述计算机指令时执行所述的一种注释宏病毒组原始测序数据短读序列的方法的步骤。
25.本发明的有益效果是:
26.(1)在本发明的一示例性实施例中,实现了宏病毒组原始数据注释的本地化、轻便化,便于不具备生物信息学背景知识的研究人员使用;在当今大型机尚未普及的情况下,解决了等待第三方公司分析周期长、收费高等问题,为宏病毒组原始数据注释提供了一种快速、简便、节省成本的方法。具体地:可以在windows个人电脑上实现宏病毒组测序原始数据病毒短读序列的注释,不需在第三方公司服务器上排队,节省大量周转时间,便于满足“快速检测”的需求;召回率非常高:对两个viral mock community(vmc)(bioproject id prjna431646and prjna395784)的召回率为100%,与nadim j ajami的virmap一致,便于满足“全面检测”的需求;操作简便全自动,便于没有生物信息学背景的科研人员或临床工作者使用;自动按照注释的病毒种提取短读序列,便于后续研究;计算资源需求低,大多windows个人电脑上可以实现分析,在配置为2.8ghz 2核intel(r)celeron(r)cpu g3900、4gb内存的windows7电脑上分析10gb规格的宏病毒组测序数据仅需1.7小时。
27.(2)在本发明的又一示例性实施例中,在使用之前,会检测特定目录下是否存在用户提供的fasta数据集:如果不存在,则使用自带的refseq病毒数据库;如果存在,则对用户提供的fasta数据集构建新的blastn索引。采用该种方式,便于数据库的更新,同时方便用户利用自制的数据集构建数据库。
28.(3)在本发明的又一示例性实施例中,脚本库包括数据库自检脚本、毒株名添加脚本和短读序列提取脚本,采用脚本实现多个步骤的匹配,其作用在于更好地实现自动化处理,调理更清晰。
29.(4)在本发明的又一示例性实施例中,质量差为比对长度小于第一长度和/或相似度小于第一阈值,其效果在于过滤掉匹配度较差的比对结果。
附图说明
30.图1为本发明一示例性实施例中提供的方法流程图。
具体实施方式
31.下面结合附图对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
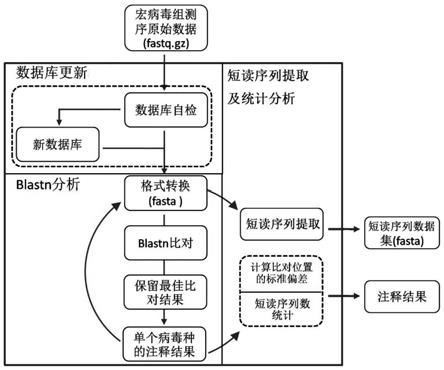
32.在本发明的描述中,需要说明的是,属于“中心”、“上”、“下”、“左”、“右”、“竖直”、“水平”、“内”、“外”等指示的方向或位置关系为基于附图所述的方向或位置关系,仅是为了便于描述本发明和简化描述,而不是指示或暗示所指的装置或元件必须具有特定的方位、以特定的方位构造和操作,因此不能理解为对本发明的限制。
33.在本发明的描述中,需要说明的是,除非另有明确的规定和限定,属于“安装”、“相连”、“连接”应做广义理解,例如,可以是固定连接,也可以是可拆卸连接,或一体地连接;可以是机械连接,也可以是电连接;可以是直接相连,也可以通过中间媒介间接相连,可以是两个元件内部的连通。对于本领域的普通技术人员而言,可以具体情况理解上述术语在本发明中的具体含义。
34.在本技术使用的术语是仅仅出于描述特定实施例的目的,而非旨在限制本技术。在本技术和所附权利要求书中所使用的单数形式的“一种”、“所述”和“该”也旨在包括多数形式,除非上下文清楚地表示其他含义。还应当理解,本文中使用的术语“和/或”是指并包含一个或多个相关联的列出项目的任何或所有可能组合。
35.应当理解,尽管在本技术可能采用术语第一、第二、第三等来描述各种信息,但这些信息不应限于这些术语。这些术语仅用来将同一类型的信息彼此区分开。例如,在不脱离本技术范围的情况下,第一信息也可以被称为第二信息,类似地,第二信息也可以被称为第一信息。取决于语境,如在此所使用的词语“如果”可以被解释成为“在
……
时”或“当
……
时”或“响应于确定”。此外,属于“第一”、“第二”仅用于描述目的,而不能理解为指示或暗示相对重要性。
36.此外,下面所描述的本发明不同实施方式中所涉及的技术特征只要彼此之间未构成冲突就可以相互结合。
37.宏病毒组测序原始数据是一种压缩的文本文件,其内容包含被测文库的核酸序列以及测序质量的分值,需要进一步将核酸序列注释到数据库中以进行病毒种的鉴定以及后续定量分析。在现有技术中,由于很多微生物研究人员或者临床工作者不具备生物信息学知识,很多基层实验室不具备服务器,像metawrap、drvm、virmap等大多序列比对程序需要在linux平台上运行,且需要较高的计算资源,即目前宏病毒组测序数据的注释和提取无法在windows平台上实现,科研人员可能会花费数周时间等待第三方公司出具物种鉴定报告,具有周期长、费用高的弊端,无法做到“利用高通量测序快速鉴定微生物种”的能力,极大阻碍科研进展,拖慢临床检测速度。因此下述示例性实施例将针对上述问题进行阐述:
38.参见图1,图1示出了本发明的一示例性实施例提供一种注释宏病毒组原始测序数据短读序列的方法的流程图,基于windows系统,包括以下步骤:
39.s02:获取原始测序数据,对原始测序数据进行解压并提取短读序列,生成短读序列数据集,调用blastn程序将数据集比对到数据库中;
40.s04:保留每条短读序列的最佳比对结果,并将最佳比对结果中比对质量差的结果去除,然后按照基因id添加病毒种名称;
41.s06:基于脚本库,统计注释到每个病毒种的短读序列数目,计算每个病毒种的短读序列在基因组上比对位置的标准偏差,并按照注释的病毒种提取对应的短读序列序列,输出fasta数据集。
42.其中,在步骤s02中,所述原始测序数据为fastq.gz格式((一种gzip压缩的文本文件),所述解压包括解压和格式转换即从fastq.gz到fasta,在提取短读序列(不提取测序质量的分值)后生成短读序列数据集(fasta格式),最后调用blastn程序将数据集比对到数据库中;
43.之后在步骤s04中,由于一条短读序列可能会存在多个比对结果,因此在该示例性
实施例中,保留每条短读序列的最佳比对结果,并将最佳比对结果中比对质量较差的结果去除(需要说明的是,即便是最佳的比对结果,比对质量(即比对长度和比对相似性)也可能很低,如果不去除比对质量较低的结果,会增加鉴定的假阳性率。比如人类基因组11q染色体的133106
–
133255bp与bean58058病毒的基因组存在一定相似性,可能会注释为该病毒,但通常比对质量很低),然后按照基因id添加病毒种名称(毒株名称)(需要说明的是,gene id是ncbi给定的唯一的毒株身份id,是一串数字;blastn程序比对后输出的结果中只包含gene id,不包含病毒毒株名称,因此需要单独添加毒株名称)。
44.最后在步骤s06中,基于脚本库,统计注释到每个病毒种的短读序列数目,计算每个病毒种的短读序列在基因组上比对位置的标准偏差(需要说明的是,如果短读序列均匀地分散在参考基因组上,则比对位置的标准偏差会很大;如果短读序列丰度较高且比对位置的标准偏差较小,说明这些短读序列可能被错误地比对到了某个高度同源的片段上。计算标准偏差的目的是为了帮助使用者判定结果是否为假阳性。但是,该方法仅计算标准偏差,而并不自动判定并去除假阳性,是因为实际的检测较为复杂,比如某些重要病原(如禽流感)可能被某些野生动物少量携带而不大量繁殖,因此测得的短读序列较少,或者分节段的双链rna病毒(如轮状病毒)在建库的过程中容易加接头失败,导致只有几个短读序列被测得
…
这些低丰度物种可能会被当作污染而忽视。因此,本示例性实施例的方法不进行假阳性的判定和筛除,旨在保留更多的信息,通过汇报平均比对相似度、短读序列数和比对位置的标准偏差来保留更多信息,以满足后续“全面检测、应检尽检”的需求),并按照注释的病毒种提取对应的短读序列序列,输出fasta数据集。便于后续引物设计,也可以使用seqman这类经典程序进行短读序列的组装。
45.其中,在本示例性实施例中,该方法实现了在windows系统中的完全自动的注释宏病毒组测序短读序列的管道,使用shell和perl编写,在windows上通过gitbash提供全部所需环境(即shell和perl环境可由git for windows程序提供),从而解决现有技术中无法在windows电脑上运行,管道中diamond、bowtie、virfinder等程序必须在linux平台上运行,且不便于没有计算机背景知识的研究人员使用的问题。也就是说,本示例性实施例实现了宏病毒组原始数据注释的本地化、轻便化,便于不具备生物信息学背景知识的研究人员使用;在当今大型机尚未普及的情况下,解决了等待第三方公司分析周期长、收费高等问题,为宏病毒组原始数据注释提供了一种快速、简便、节省成本的方法。
46.其中,需要说明的是,二代测序诞生至今已有17年,然而尚没有软件在windows平台实现宏病毒组测序数据的注释,原因是无桌面的linux系统比windows具有更高的性能。平台转换的问题却一直未解决。由于大部分用户使用windows系统,因此在windows系统中实现宏病毒组原始数据的注释,虽不具备技术方面的区别或优势,但具有应用上的必要性。现有技术中的管道调用的程序有很多,比如使用cutadapt和trimmomatic软件进行接头序列的去除,此步骤对contigs的组装质量有显著提高,而对短读序列的局部比对搜索(basic local alignment search)无影响,而本示例性实施例的管道不基于组装,因此无需进行这一步骤。现有技术的管道使用soap.coverage程序来统计丰度,而本示例性实施例的方法直接对blastn结果中比对到各个病毒基因组的短读序列数进行统计即可得到丰度,冗余步骤较少。
47.因此,本示例性实施例具有以下优点:1.可以在windows个人电脑上实现宏病毒组
测序原始数据病毒短读序列的注释,不需在第三方公司服务器上排队,节省大量周转时间,便于满足“快速检测”的需求;2.召回率非常高:对两个viral mock community(vmc)(bioproject id prjna431646 and prjna395784)的召回率为100%,与nadim j ajami的virmap一致,便于满足“全面检测”的需求;3.操作简便全自动,便于没有生物信息学背景的科研人员或临床工作者使用;4.自动按照注释的病毒种提取短读序列,便于后续研究;5.计算资源需求低,大多windows个人电脑上可以实现分析,在配置为2.8ghz 2核intel(r)celeron(r)cpu g3900、4gb内存的windows7电脑上分析10gb规格的宏病毒组测序数据仅需1.7小时。
48.需要说明的是,perl是practical extraction and report language的缩写,它是由larry wall设计的,并由他不断更新和维护,用于在unix环境下编程。perl具有高级语言(如c)的强大能力和灵活性。事实上,它的许多特性是从c语言中借用来的。与脚本语言一样,perl不需要编译器和链接器来运行代码,要做的只是写出程序并告诉perl来运行而已。这意味着perl对于小的编程问题的快速解决方案和为大型事件创建原型来测试潜在的解决方案是十分理想的。perl提供脚本语言(如sed和awk)的所有功能,还具有它们所不具备的很多功能。perl还支持sed到perl及awd到perl的翻译器。per像c一样强大,像awk、sed等脚本描述语言一样方便。而对于shell即为供用户使用的界面,shell为用户提供了输入命令和参数并可得到命令执行结果的环境。总体而言,shell是一个解释“程序”,专门用来解释执行各种命令;perl是一种脚本语言,用它写出来的脚本程序可以通过shell来解释执行。
49.git for windows专注于提供一套轻量级的本地工具集,它将git scm的完整功能集引入windows,同时为git用户提供适当的用户界面,方便用户在windows下使用git。git for windows主要提供两个工具:一个是git bash命令行,另一个git gui图形用户界面。
50.fastq格式是一种包含质量值的序列文件,其中的q为quality,一般用来存储原始测序数据,扩展名一般为fastq或者fq。下面是fastq格式常见的序列格式:第一行:以'@'开头,是这一条短读序列的名字,这个字符串是根据测序时的状态信息转换过来的,中间不会有空格,它是每一条短读序列的唯一标识符,同一份fastq文件中不会重复出现,甚至不同的fastq文件里也不会有重复;第二行:测序短读序列的序列,由a,c,g,t和n这五种字母构成,这也是真正关心的dna序列,n代表的是测序时那些无法被识别出来的碱基;第三行:以'+'开头,在旧版的fastq文件中会直接重复第一行的信息,但现在一般什么也不加(节省存储空间);第四行:测序短读序列的质量值,这个和第二行的碱基信息一样重要,它描述的是每个测序碱基的可靠程度,用asci码表示。目前绝大部分的软件都可以直接处理压缩格式,因此一般的fastq格式都是压缩格式呈现的扩展名为fq.gz,如果需要压缩或者解压缩可以进行。
51.fasta格式是一种基于文本用于表示核酸序列或多肽序列的格式。其中核酸或氨基酸均以单个字母来表示,且允许在序列前添加序列名及注释。该格式已成为生物信息学领域的一项标准。fasta格式文件的第一行是由大于号“》”(较常用)或分号“;”打头的任意文字说明,用于序列标记。从第二行开始为序列本身,只允许使用既定的核苷酸或氨基酸编码符号(参见支持代码类型)。通常核苷酸符号大小写均可,而氨基酸常用大写字母。使用时应注意有些程序对大小写有明确要求。一般每行60~80个字母。
52.blast程序包括以下五个:(1)blastp:将待查询的蛋白质序列及其互补序列一起对蛋白质序列数据库进行查询;(2)blastn:将待查询的核酸序列及其互补序列一起对核酸序列数据库进行查询;(3)blastx:先将待查询的核酸序列按六种可读框架(逐个向前三个碱基和逐个向后三个碱基读码)翻译成蛋白质序列,然后将翻译结果对蛋白质序列数据库进行查询;(4)tblastn:先将核酸序列数据库中的核酸序列按六种可读框架翻译成蛋白质序列,然后将待查询的蛋白质序列及其互补序列对其翻译结果进行查询;(5)tblastx:先将待查询的核酸序列和核酸序列数据库中的核酸序列按六种可读框架翻译成蛋白质序列,然后再将两种翻译结果从蛋白质水平进行查询。其中,本示例性实施例的步骤s02采用的是第(2)中的方式。
53.另外,现有技术中用到了组装的方法,这样算耗会比较大,不便于在大多个人电脑上运行;而在本示例性实施例中,本方法实现宏病毒组测序数据中短读序列的注释,比基于组装的注释保留更丰富的信息。有些低丰度病毒的短读序列是不连续的,这些短读序列之间没有重叠(overlaps)部分,因此无法进行组装,得到的contigs不会包含这一部分病毒。如果基于组装的方法进行注释,会损失掉这些病毒的信息,且算耗较大。现有技术中进行组装的主要目的是为了获得完整的基因(组)序列并进行后续分析,比如进行病毒序列的预测
……
与本管道目的不同。
54.更优地,在一示例性实施例中,如图1所示,所述方法还包括以下步骤:
55.s01:检测特定目录下是否存在判断是否存在blastn索引,如果不存在blastn索引则进一步判断特定目录下是否存在用户提供的fasta数据集,如果存在用户提供的fasta数据集则基于所述fasta数据集构建新的blastn索引,否则进一步判断特定目录下是否存在refseq病毒数据库,如果存在refseq病毒数据库则基于所述refseq病毒数据库构建新的blastn索引,否则报错;在存在blastn索引的情况下,完成后续注释步骤。
56.具体地,在该步骤中,所述特定目录可以为windows系统创建的/databases目录,refseq病毒数据库可以为当年的refseq病毒基因序列库(例如2021年refseq病毒基因序列库,其中可以包括全部的非噬菌体病毒);而所述对用户提供的fasta数据集构建新的blastn索引,可以采用makeblastdb程序实现。
57.其中,可以有以下几种:如果/databases目录下存在blastn索引(即为数据库文件),则直接进行下一步流程(步骤s02);如果不存在blastn索引,则程序利用自带的2021年refseq病毒基因序列数据集构建blastn索引,然后进行下一步流程(步骤s02);如果不存在blastn索引且存在用户提供的带有gene id的fasta数据集,则程序会整合这个数据集以及原有的数据集以构建blastn索引,然后进行下一步流程(步骤s02);如果既不存在任何数据集(即没有用户提供的带有gene id的fasta数据集,也没有自带的2021年refseq病毒基因序列数据集)也不存在blastn索引,则程序会报错(用户误删导致)。
58.具体地,在该步骤中,更新、自制数据库极其便利:删除/databases目录下原有的数据库并把自己的数据集(fasta格式,并带有gene id)放进该目录即可在下次运行程序的时候自动整合原有数据集。更新数据库是为了对新发病毒进行更准确的注释;自制数据库的应用场景很多,比如某些病毒(如猪星状病毒)具有丰富的遗传多样性,在研究该病毒的混合感染时,用户可以单独在ncbi上下载同种不同型毒株以自制索引,如此可以增加病毒鉴定的分辨率;比如需要单独搜寻强毒毒株的毒力基因或决定特殊表型的关键基因时,通
过自制数据库,可以大大减小算耗,增加搜索的精准性。又例如,可以将噬菌体序列的数据集直接放进存储数据库的文件夹即可完成数据库的更换,然后就可以直接对高通量测序原始数据中的噬菌体进行注释。
59.更优地,在一示例性实施例中,所述脚本库包括数据库自检脚本、毒株名添加脚本和短读序列提取脚本。
60.其中,数据库自建脚本用于对上述的步骤s01进行实现,毒株名添加脚本用于对上述的步骤s04进行实现,短读序列提取脚本用于对步骤s02实现。另外,各个脚本均为步骤s06中提取短读序列时所需;由于本示例性实施例在windows系统上实现,采用perl脚本效率高,采用脚本匹配的形式在功能上更加有条理。
61.更优地,在一示例性实施例中,所述质量差为比对长度小于第一长度和/或相似度小于第一阈值。
62.其中,在其中一示例性实施例,所述比对长度小于第一长度的具体条件为比对长度《80bp,而相似度小于第一阈值的具体条件为相似度小于80%;长度和相似度可以采用“和/或”的形式实现。
63.需要说明的是,二代测序的短读序列长度普遍为100~250bp。根据经验,短读序列的比对长度《80bp且核苷酸相似性低于80%的比对结果通常为假阳性;相似度的计算方式为:参与比对的区段中,完全一致的碱基数/比对区段的长度
×
100%。
64.与上述示例性实施例具有相同的发明构思,本发明的又一示例性实施例提供一种注释宏病毒组原始测序数据短读序列的系统,基于windows系统,包括:
65.blastn分析模块:用于获取原始测序数据,对原始测序数据进行解压并提取短读序列,生成短读序列数据集,调用blastn程序将数据集比对到数据库中;保留每条短读序列的最佳比对结果,并将最佳比对结果中比对质量差的结果去除,然后按照基因id添加病毒种名称;
66.短读序列提取及统计分析模块:用于基于脚本库,统计注释到每个病毒种的短读序列数目,计算每个病毒种的短读序列在基因组上比对位置的标准偏差,并按照注释的病毒种提取对应的短读序列序列,输出fasta数据集。
67.与所述方法示例性实施例类似的,实现了在windows系统中的完全自动的注释宏病毒组测序短读序列的管道,使用shell和perl编写,在windows上通过gitbash提供全部所需环境(即shell和perl环境可由git for windows程序提供),从而解决现有技术中无法在windows电脑上运行,管道中diamond、bowtie、virfinder等程序必须在linux平台上运行,且不便于没有计算机背景知识的研究人员使用的问题。也就是说,本示例性实施例实现了宏病毒组原始数据注释的本地化、轻便化,便于不具备生物信息学背景知识的研究人员使用;在当今大型机尚未普及的情况下,解决了等待第三方公司分析周期长、收费高等问题,为宏病毒组原始数据注释提供了一种快速、简便、节省成本的方法。
68.因此,本示例性实施例具有以下优点:1.可以在windows个人电脑上实现宏病毒组测序原始数据病毒短读序列的注释,不需在第三方公司服务器上排队,节省大量周转时间,便于满足“快速检测”的需求;2.召回率非常高:对两个viral mock community(vmc)(bioproject id prjna431646and prjna395784)的召回率为100%,与nadim j ajami的virmap一致,便于满足“全面检测”的需求;3.操作简便全自动,便于没有生物信息学背景的
科研人员或临床工作者使用;4.自动按照注释的病毒种提取短读序列,便于后续研究;5.计算资源需求低,大多windows个人电脑上可以实现分析,在配置为2.8ghz 2核intel(r)celeron(r)cpu g3900、4gb内存的windows7电脑上分析10gb规格的宏病毒组测序数据仅需1.7小时。
69.对应地,在一示例性实施例中,所述系统还包括:
70.数据库更新模块,用于检测特定目录下是否存在用户提供的fasta数据集:如果不存在,则使用自带的refseq病毒数据库作为数据库;如果存在,则对用户提供的fasta数据集构建新的blastn索引作为数据库。
71.对应地,在一示例性实施例中,所述脚本库包括数据库自检脚本、毒株名添加脚本和短读序列提取脚本。
72.对应地,在一示例性实施例中,所述质量差为比对长度小于第一长度和/或相似度小于第一阈值。
73.与上述示例性实施例具有相同的发明构思,本发明的又一示例性实施例提供一种存储介质,其上存储有计算机指令,所述计算机指令运行时执行所述的一种注释宏病毒组原始测序数据短读序列的方法的步骤。
74.与上述示例性实施例具有相同的发明构思,本发明的又一示例性实施例提供一种装置,包括存储器和处理器,所述存储器上存储有可在所述处理器上运行的计算机指令,所述处理器运行所述计算机指令时执行所述的一种注释宏病毒组原始测序数据短读序列的方法的步骤。
75.基于这样的理解,本实施例的技术方案本质上或者说对现有技术做出贡献的部分或者该技术方案的部分可以以软件产品的形式体现出来,该计算机软件产品存储在一个存储介质中,包括若干指令用以使得一台计算机设备(可以是个人计算机,服务器,或者网络设备等)执行本发明各个实施例所述方法的全部或部分步骤。
76.存储器用于存储各种类型的数据以支持在该装置的操作,这些数据例如可以包括用于在该装置上操作的任何应用程序或方法的指令,以及应用程序相关的数据,例如联系人数据、收发的消息、图片、音频、视频等等。该存储器可以由任何类型的易失性或非易失性存储设备或者它们的组合实现,例如静态随机存取存储器(static random access memory,简称sram),电可擦除可编程只读存储器(electrically erasable programmable read-only memory,简称eeprom),可擦除可编程只读存储器(erasable programmable read-only memory,简称eprom),可编程只读存储器(programmable read-only memory,简称prom),只读存储器(read-only memory,简称rom),磁存储器,快闪存储器,磁盘或光盘。
77.显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定,对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其他不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引申出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1