一种前列腺癌的诊断模型构建方法
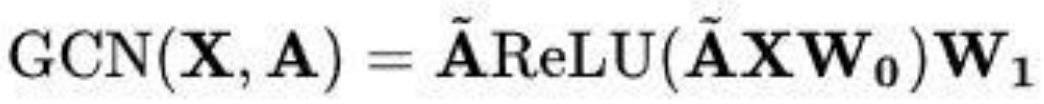
1.本发明涉及医学领域,尤其涉及一种前列腺癌的诊断模型构建方法。
背景技术:
2.前列腺癌(porstate cancer,pca)是西方国家第二大最常在男性中被诊断为恶性的肿瘤。根据世界卫生组织(world health organization,who)国际癌症研究机构的数据,截至2012年全世界约有110万男性被诊断出患有前列腺癌,占男性确诊的所有癌症的15%。在我国,根据2019年1月国家癌症中心发布的最新一期全国癌症统计数据,前列腺癌发病率近年来有明显的上升趋势,在男性中排在第6位。pca被认为是一种异质性疾病,多个基因和细胞通路共同参与了pca的发生与发展。细胞的表观遗传突变导致的肿瘤可能无法控制地生长和繁殖。
技术实现要素:
3.有鉴于此,本发明的目的之一是提供一种前列腺癌诊断模型,该模型可以实现对前列腺癌的预测。
4.本发明通过以下技术手段解决上述技术问题:
5.一种前列腺癌的诊断模型的构建方法,包括以下步骤:
6.step1)获取pca(前列腺癌)的基因表达谱数据;
7.step2)对pca的基因表达谱数据进行差异表达谱分析,筛选出pca中的差异基因;
8.step3)针对pca中的差异基因通过机器学习方法中的gae(graph autoencoder)算法筛选关键基因;
9.step4)对gae的计算结果,通过ppi分析得到关键基因中的10个高表达基因和6个低表达基因;
10.step5)通过单因素回归分析和多因素回归分析建立预后模型;
11.step6)根据预后模型参数构建pca的诊断模型;
12.step7)对pca诊断模型进行验证。
13.基于pca差异表达基因,通过机器学习中的gae的方法进行分析,并筛选出pca中的高表达基因为:ube2c、ccnb1、top2a、tpx2、cenpm、kiaa0101、f5、apoe、npy和trim36,低表达基因为:myh11、flna、acta2、myl9、tagln和actg2。
14.通过单因素cox比例风险模型找出与pca预后相关的关键基因,再通过多因素cox比例风险模型构建一个基于4个基因的诊断模型。述诊断模型通过以下公式计算:
15.预后风险指标=(0.3153
×
top2a基因表达水平)+(0.2987
×
ube2c基因表达水平)+(-0.7064
×
myl9基因表达水平)+(-0.4628
×
flna基因表达水平)
16.本发明的有益效果:
17.本发明发现并验证了由4个与pca预后相关的关键基因构成的诊断模型。另外,通过整合多组学数据库验证构建预测模型的关键基因,本发明获得的结果为pca生物标志物
的研究提供了新方向,同时也为pca患者的个性化精准治疗提供了新的可能性。
附图说明
18.下面结合附图和实施例对本发明作进一步的阐述;
19.图1为gse6919和gse30174两个数据集的表达谱;
20.图2为gse6919和gse30174数据集差异表达谱分析结果;
21.图3为gae算法筛选出的关键基因;
22.图4为通过ppi分析后得到的显著上调和显著下调基因;
23.图5为geo训练集中高低风险基因表达;
24.图6为geo训练集中roc曲线;
25.图7为预测模型的多因素cox分析;
26.图8为预测模型与年龄、病理分期的多因素cox分析;
27.图9为预测模型的roc曲线;
28.图10为通过gepia数据库验证关键基因;
29.图11为通过oncomie数据库验证关键基因;
30.图12为通过getx数据库验证关键基因;
31.图13为通过human proteinatlas数据库验证关键基因。
具体实施方式
32.以下结合具体实验对本发明作详细的说明:
33.本发明的一种前列腺癌的诊断模型构建方法,具体包括以下步骤:
34.步骤一:数据收集与分析
35.1)收集患者数据
36.从gene expression omnibus(geo)数据库中选择gse6919和gse30174两个数据集作为训练数据集。
37.geo数据库是一个公共的基因组数据数据库,其中的数据都是来自于公开发表的论文中的内容。该数据库创建于2000年,收录了世界各国研究机构提交的高通量基因表达数据,也就是说只要是目前已经发表的论文,论文中涉及到的基因表达检测的数据都可以通过这个数据库中找到。因此,基于该数据库作为数据来源具有较高的可信度。
38.本发明选择了gse6919和gse30174两个数据集作为机器学习的训练数据源。gse6919数据集是基于agilent gpl92、gpl93和gpl8300平台(affymetrix human genome u95 version 2array)的,由federico alberto monzon于2018年提交。gse30174数据集共有504个样本,其中包括233个正常前列腺组织和271个转移性前列腺肿瘤。gse30174由jennifer barb于2019提交。训练集的表达谱数据如图1所示。
39.使用gse16560数据集作为验证数据集。gse16560数据集包含80个样本,包括10个健康外周血和70个非转移性前列腺肿瘤。gse16560作为验证数据集基于gpl5474平台(用于dasl的人类6k转录信息基因组),由andrea sboner于2013年提交,包含281个样本,包括由不同gleason score排序的原发性前列腺肿瘤。
40.2)在前列腺癌中筛选差异表达基因
41.为了筛选pca中的差异基因,本发明使用r语言中的limma软件包,对gse6919和gse30174数据集差异表达谱分析,从中筛选了6269个差异基因。筛选标准为(false discovery rate,fdr)《0.05和|log2|(fold change,fc)|》1.5。
42.gse6919和gse30174数据集差异表达谱分析结果如图2所示。
43.进一步,go分析结果显示这些差异基因在生物过程(bp)中显着富集,包括信号转导、rna聚合酶ii启动子转录的正调控。细胞组分(cc)分析表明,这些差异基因在细胞质囊泡膜、膜的整体成分和质膜中显着富集。对于分子功能(mf),这些差异基因富含蛋白质结合、蛋白质同二聚化活性和钙离子结合。
44.kegg分析结果表明,所有上调基因在扩张型心肌病、肥厚型心肌病(hcm)、ecm-受体相互作用、致心律失常性右心室心肌病(arvc)、粘着斑和tgf-β信号通路中均显着富集。
45.3)使用gae机器学习算法进一步筛选pca关键基因
46.gae(图自动编码器,graphauto encoder)是一种无监督的学习模型。gae的相关变量如下:
47.图用g可以通过g=(v,e)来表示,其中v表示节点的集合,e表示边的集合。
48.a:表示邻接矩阵
49.d:表示度矩阵,文中假设对角线上元素均为1
50.n:表示节点数
51.d:表示节点的特征维度
52.x{\epsilon}{\mathbb{r}}^{n*d}x∈rn*d:表示节点的特征矩阵
53.f:表示embedding维度
54.z{\epsilon}{\mathbb{r}}^{n*f}z∈rn*f:表示节点的embedding
55.gae的编码过程是,gae使用gcn作为encoder,来得到节点的latent representations(或者说embedding),这个过程可如下公式表示:
56.z=gcn(x,a)
57.将gcn视为一个函数,然后将x和a作为输入,输入到gcn函数中,输出z{\epsilon}{\mathbb{r}}^{n*f}z∈rn*f,z代表的就是所有节点的latent representations,或者说embedding。gcn的函数定义如下:
[0058][0059]
从公式上可以看出,整个encoder只有两层,每一层采用的均是切比雪夫多项式的一阶近似作为卷积核处理数据。从中可以看出除了初始的输入x也就是表示节点的特征矩阵,剩下的参数均是需要学习的对象。简言之,这里gcn就相当于一个以节点特征和邻接矩阵为输入、以节点embedding为输出的函数,目的只是为了得到embedding。
[0060]
gae的编码过程是,gae采用inner-product作为解码来重构(reconstruct)原始的图:
[0061][0062]
所得就是重构出来的邻接矩阵,根据此邻接矩阵和原图的信息特征就可以构造损失函数。
[0063]
gae的损失函数是,邻接矩阵决定了图的结构,应该使重构出的邻接矩阵与原始的
邻接矩阵尽可能的相似。因此,gae在训练过程中,采用交叉熵作为损失函数:
[0064][0065]
上式中,y代表邻接矩阵a中某个元素的值(0或1),\hat{y}y^代表重构的邻接矩阵\hat{a}a^中相应元素的值(0到1之间)。从损失函数可以看出来,希望重构的邻接矩阵(或者说重构的图),与原始的邻接矩阵(或者说原始的图)越接近、越相似越好。
[0066]
在得到pca的差异基因后,本发明使用tensorflow实现gae算法,这将从6269个差异基因中筛选出关键基因。gae使用编码器学习网络嵌入以提取网络嵌入,并使用解码器来执行网络嵌入以通过邻接矩阵保留节点的拓扑信息:
[0067][0068]
其中v1,v2∈v,并且count(
·
)函数返回节点v和或节点u在随机采样中共同出现/出现的频率分布。
[0069]
通过gae算法筛选出的关键基因如图3所示。
[0070]
在得到关键基因后,将所有关键基因都上传到string数据库进行ppi(protein-protein interaction)分析(图4)。string数据库共包含14094个器官、6.76千万个蛋白以及超过20亿个交互。这为关键基因间的交互研究提供了重要的基础。本发明的ppi结果共有6475个节点以拓扑形式呈现,由gae生成的前100个基因中,显着上调的基因为:ube2c、ccnb1、top2a、tpx2、cenpm、f5、apoe、npy和trim36;显着下调的基因为:myh11、flna、acta2、myl9、tagln和actg2。
[0071]
步骤二:模型构建及模型验证
[0072]
4)预测模型的构建和验证
[0073]
首先,采用单变量cox分析来研究患者os与每个关键基因的表达水平之间的关系。分析工具采用r语言中的survminer r。在单变量cox回归分析中p值《0.01的筛选条件被认为是显着的。
[0074]
其次,进行多变量cox比例风险分析,以评估多个基因作为影响患者生存的独立预后因素的贡献。
[0075]
最后,采用逐步法选择优化模型。通过使用多因素cox回归的系数作为权重,构建pca风险评分预测模型。风险评分计算如下:
[0076]
风险评分=∑风险基基因i×
基因表达量i[0077]
本发明构建的预后风险指标=(0.3153
×
top2a基因表达水平)+(0.2987
×
ube2c基因表达水平)+(-0.7064
×
myl9基因表达水平)+(-0.4628
×
flna基因表达水平)
[0078]
本发明中使用geo训练集构建的高低风险基因表达结果如图5所示。
[0079]
对于本发明所构建的预后风险模型的性能评估,则是使用roc曲线来评估预测性能,并使用geo数据库中的gse16560数据集来验证。
[0080]
geo训练集的roc曲线如图6所示。
[0081]
5)验证预测模型与临床相关信息之间的独立性
[0082]
采用单变量和多变量cox回归分析评估tcgaprad队列和gse 16560队列四基因预
后模型的独立预测价值。并且通过单变量cox回归分析分析了具有格里森评分和病理分期的临床信息。由于年龄和格里森评分几乎达到统计学显着性,我们将年龄、格里森评分和预后模型结合到多变量cox回归分析中(图7),分析表明多变量cox回归分析结果显示预后模型与os无关。
[0083]
图7的cox多因素分析表明,本发明构建的风险模型p=0.0073,p《0.01,与影响pca诊断的其他因素具有明显的差异。
[0084]
此外,gse16560数据集用于评估预后模型的预测价值(图8)。使用最佳风险临界值将gse16560数据集中的280名患者分为高风险组(n=190)和低风险组(n=90)。用于预后模型的生存预测的时间依赖性roc分析获得了1年时0.69、3年时0.58和5年时0.61的auc(图9)。
[0085]
6)验证构建预测模型4个关键基因
[0086]
分别使用gepia、oncomine、getx和human proteinatlas数据库对构建预测模型的4个关键基因进行验证。
[0087]
gepia数据库可以用于验证本预测模型中4个关键基因在前列腺癌中的表达是否显著(图10)。
[0088]
oncomine数据库可以用于验证本预测模型中4个关键基因在多种肿瘤中的表达情况(图11)。
[0089]
getx数据库可以用于验证本预测模型中4个关键基因在正常组织中的表达情况(图12)。
[0090]
human proteinatlas数据库可以用于验证本预测模型中4个关键基因在病理中的表达情况(图13)。
[0091]
以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的宗旨和范围,其均应涵盖在本发明的权利要求范围当中。本发明未详细描述的技术、形状、构造部分均为公知技术。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1