用于脂肪肝病的修饰的吻素受体激动剂
用于脂肪肝病的修饰的吻素受体激动剂
1.相关申请的交叉引用
2.本技术要求2020年4月8日提交的美国临时申请序列号63/007,071的权益。该申请的全部内容通过援引并入在此,就好像在本文中完全阐述一样。
3.背景
1.技术领域
4.本发明涉及医学领域,并且特别涉及减少受试者肝脏中的脂肪的方法。该方法包括向受试者给药tak-448或其任何盐;tak-448的保守变体或其任何盐;吻素-10或其任何盐;或吻素-10的保守变体或其任何盐。
2.
背景技术:
5.肝脏是参与脂质代谢的主要器官。血脂异常会导致代谢紊乱,诸如非酒精性脂肪性肝病(nafld),这已成为影响全世界约10亿人的日益严重的公共卫生问题。在美国,nafld是影响超过1200万成年人和800万儿童的全国性流行病,相关的年度医疗费用为1030亿美元。
6.nafld是全世界肝病的主要原因。nafld的特征是肝脏脂肪积累(脂肪变性),导致产生细胞毒性脂质氧化副产物,进而发展为伴有肝细胞损伤的慢性炎症状态,定义为非酒精性脂肪性肝炎(nash)。随着疾病的进展,一部分患者将发展为纤维化、肝硬化和肝功能衰竭或肝细胞癌(hcc)。nafld/nash已取代丙型肝炎成为未来十年最常见的肝移植适应症。因此,nafld涵盖了一系列病理病症,包括脂肪变性(脂肪肝)、非酒精性脂肪性肝炎(nash)以及纤维化和肝硬化(肝脏过度的不可逆瘢痕化)。
7.吻素(kisspeptin,kp)是kiss1基因的肽产物,是g蛋白偶联受体吻素1受体(kiss1r)的内源配体。kp/kiss1r信号传导系统在中枢(在脑中)和外周表达,其中其在生殖和新陈代谢中起主要作用。事实上,在肥胖的遗传模型(db/db和ob/ob小鼠)中发现肝脏kiss1表达增加。虽然kiss1和kiss1r在肝脏中表达,但肝脏kiss1r信号传导在调节脂肪生成中的作用尚不清楚。
8.nafld的流行反映了影响约1亿美国人的肥胖和ii型糖尿病的增加。然而,用于治疗ii型糖尿病的药物未被批准用于治疗nafld。除了在疾病早期有益的减肥和减肥手术外,目前还没有fda批准的药物来治疗nafld/nash。fda对行业的指导表示:“fda认为,确定将阻止或逆转nash和nafld或减缓其进展的疗法将解决未满足的医疗需求。”参见fda.gov(2018年12月)。随着肥胖率持续上升,美国nafld相关肝病和死亡率将上升。到2026年,nash治疗和肝硬化药物市场的全球市场预计将分别达到18.3b美元和66.01b美元。
9.因此,迫切需要开发用于nafld/nash的新治疗。
技术实现要素:
10.本发明涉及在nafld或nash中使用tak-448的再利用。在这项研究中,使用高脂肪
饮食诱导的nafld临床前小鼠模型来证明kiss1r的肝脏敲除促进了脂肪变性。此外,在肥胖的野生型糖尿病小鼠中输注kp类似物(tak-448)防止疾病进展。在机制上,kp信号传导被证明除了抑制过氧化物酶体增殖物激活受体-γ(pparγ)信号通路外,还通过激活amp活化蛋白激酶(ampk)以及通过促进线粒体中脂肪的β氧化或分解来负调节从头脂肪生成(脂肪形成)。这项研究提供了直接证据,表明针对kiss1r的药理学和遗传干预可以防止nafld的发展并限制其进展为nash和纤维化。
11.特别地,本发明涉及在有此需要的受试者中治疗或预防nafld/nash的方法,其包括向所述受试者给药治疗有效量的tak-448。优选地,受试者是nafld患者或nash患者。可以通过任何途径给药,但优选皮下。
12.本发明的某些实施方式包括减少有此需要的受试者的肝脏中脂肪的方法,其包括向所述受试者给药治疗有效量的选自以下的化合物:tak-448及其任何盐;tak-448的保守变体及其任何盐;吻素-10及其任何盐;以及吻素-10的保守变体及其任何盐。受试者优选患有脂肪肝和/或选自酗酒、肝炎、非酒精性脂肪肝病(nalfd)或非酒精性脂肪性肝炎(nash)的病症;最优地选受试者患有非酒精性脂肪肝病(nalfd)或非酒精性脂肪性肝炎(nash)。
13.本发明的一些实施方式涉及一种方法,其进一步包括向受试者给药治疗量的第二化合物,所述第二化合物选自akr-001、aramchol、asc40、azd7687、bio89-100、bms-986036、卡格列净(canagliflozin)、cenicriviroc、昔洛法克索(cilofexor)、达格列净(dapagliflozin)、edp-305、elafibranor、恩利卡生(emricasan)、eyp001a、非诺贝特(fenofibrate)、非索考司他(firsocostat)、gr-md-02、ionis-dgat2
rx
、伊格列净(ipragliflozin)、利可格列净(licogliflozin)、鲁格列净(luseogliflozin)、ly3202328、met409、二甲双胍(metformin)、mgl-3196、mk-4074、msdc-0602k、mt-3995、ngm282、尼度法克索(nidufexor)、pf-05221304、pf-06835919、pf-06865571、pf-06882961、px-102、pxl770、pxs-5153a、司隆色替(selonsertib)、辛妥珠单抗(simtuzumab)、simaglutide、tern-101、tern-201、troifexor、维生素d、维生素e和vk2809。
14.给药方法可以是皮下、静脉内或口服。
附图说明
15.图1a至图1j。tak-448在nafld的临床前小鼠模型中减少肝脏中的脂肪积累(脂肪变性)并改善代谢谱。图1a是对于高脂肪饮食(hfd)饲喂的对照(载剂、磷酸盐缓冲盐水或pbs)和tak-448治疗的小鼠肝脏(5只小鼠/组),苏木精和伊红染色的小鼠肝脏的组织学照片,其中指示(黑色箭头),显示了脂肪变性(肝脏中的小白色区域),以及指示(灰点)的油红o染色的肝脏脂质。与hfd饲喂的同窝崽对照相比,tak-448治疗降低了hfd诱导的肝脂肪变性。图1b至图1g显示了tak-448在降低终点肝脏甘油三酯(tg)(图1b)、终点血液甘油三酯(图1c)、血液游离脂肪酸(ffa)(图id)、终点体重(图1e)和动物外周脂肪积累(图1f(附睾白色脂肪组织或ewat)和图1g(腹股沟白色脂肪组织或iwat))中的有益效果。终点定义为小鼠安乐死后。图1h至图1j显示,tak-448治疗预防空腹血糖升高(图1h),预防葡萄糖不耐受(改善糖尿病),如使用葡萄糖耐量试验(gtt)确定的(图ii),并且在使用胰岛素耐量试验(itt)中预防胰岛素抵抗(图1j)。
16.图2a至图2c。图2a和图2b示出了在用载剂(veh、pbs)或takeda-448(tak,0.3nmol/
h)治疗之前野生型c57bl6/j小鼠的体重(图2a)和空腹血糖(图2b)。图2c显示治疗后4周,用tak治疗的小鼠或对照的食物摄取没有差异,如通过临床实验动物监测系统(clams)测量的;rd/hfd 10周;5只小鼠/组。示出了平均值
±
sem,student未配对t检验,与veh对照相比,*p《0.05。
17.图3a至图3c示出了rd/hfd(10周)饲喂的对照和tak治疗的小鼠(治疗后4周)中的脂肪肝标志物血清酶丙氨酸氨基转移酶(alt)水平(图3a)、血清胆固醇(图3b)和血清甘油(图3c)水平。与hfd饲喂的对照相比,tak-448的给药降低了hfd饲喂小鼠中的alt、胆固醇和甘油水平。
18.图4a和图4b示出了clams分析,其显示了载剂(veh、pbs)对照和tak-448治疗的小鼠(治疗后4周,rd/hfd 10周,5只小鼠/组)中的热量消耗(图4a)和呼吸交换率(rer)(图4b)。
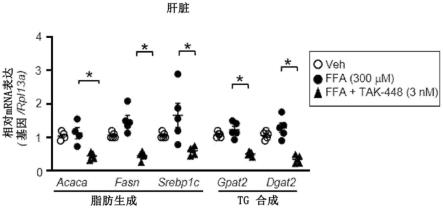
19.图5a和图5b。图5a示出了hfd肝脏样品中通过rt-qpcr的指示基因的相对mrna表达,其相对于rpl13a mrna表达进行归一化。tak-448治疗降低了调节肝脏中脂肪生产(甘油三酯合成和脂肪生成)的关键基因的水平。图5b是代表性蛋白质印迹,显示了hfd肝脏小鼠中指示蛋白质的表达。tak-448治疗降低了肝脏中脂肪生产的关键调节子的蛋白质水平。
20.图6a至图6c示出了图5b中所示的蛋白质印迹的光密度分析。
21.图7是显示pampk和总ampk的蛋白质印迹。tak-448治疗激活关键能量传感器ampk,其然后抑制脂肪形成。
22.图8a和图8b分别示出了图7和图5b中所示蛋白质印迹的光密度分析。
23.图9a和图9b示出了来自用veh(对照)或tak治疗的小鼠的hfd肝脏样品中通过rt-qpcr的炎症标志物(图9a)以及纤维化和氧化应激标志物(图9b)的相对mrna表达,其相对于rpl13a mrna表达进行归一化。tak-448治疗降低了调节肝脏中炎症和纤维化的关键基因的水平。示出了平均值
±
sem。student未配对t检验,与veh对照相比,*p《0.05。
24.图10是关于tak-448治疗对肝甘油三酯合成和从头脂肪生成的关键调节子的表达的抑制作用的图。
25.图11是关于tak-448治疗对肝炎症的关键调节子的表达的抑制作用的图。
26.图12是关于tak-448治疗对肝纤维化的关键调节子的表达的影响的图。
27.图13是关于tak-448治疗对调节脂肪的线粒体和过氧化物酶体β-氧化的肝基因表达的影响的图。
28.图14a至图14d。图14a是示出了hfd饲喂的对照(ctrl)和肝脏kiss1r敲除(lko)小鼠的体重(bw)随时间的图。图14b是示出了对照和lko小鼠的终点体重的条形图。图14c是hfd小鼠(ctrl和lko)的照片。图14d是示出了对照和lko小鼠在4天平均的每日食物摄取的条形图。
29.图15a和图15b是苏木精和伊红染色的小鼠肝脏的一组显微照片,显示了具有高脂肪(图3a)和正常(图15)饮食的ctrl和lko(hdf和rd)小鼠的脂肪变性。图15b中的方框区域在下方放大。
30.图16是示出了ctrl和lko(rd和hfd)小鼠中的肝脏甘油三酯的条形图。与同样hfd饲喂的对照相比,hfd饲喂的lko小鼠肝脏甘油三酯升高。
31.图17示出了饲喂正常饮食(rd)或高脂肪饮食(hfd)8周的c57bl6雄性小鼠中通过
rt-qpcr的kiss1和kiss1r的相对mrna表达,其相对于rpl13a mrna表达进行归一化。hfd诱导肝脏中的kiss1和kiss1r表达。
32.图18a至图18d。图18a示出了来自ctrl和显示kiss1r的敲低的lko小鼠的hfd肝脏中通过rt-qpcr的指示基因的相对mrna表达。与对照相比,lko小鼠的肝脏中缺乏kiss1r表达。示出了平均值
±
sem,student未配对t检验,与对照相比,*p《0.05。图18b示出了饲喂正常饮食(rd)的ctrl和lko小鼠的体重。维持rd的小鼠体重没有变化。图18c和图18d示出了显示ctrl和lko小鼠的呼吸交换率(rer)(图18c)和走动活动(图18d)的clams分析。*p《0.05;单向方差分析,然后是dunnett事后检验。
33.图19a和图19b。图19a示出了通过clams评估的热量消耗,反映了hfd lko小鼠的较低代谢。图19b显示,与hfd饲喂的ctrl相比,hfd饲喂的lko小鼠中肝病的标志物肝酶丙氨酸氨基转移酶(alt)的血清水平增加。
34.图20a至图20d示出了ctrl和lko小鼠中腓肠肌(图20a)、胫骨前肌(图20b)、附睾白色脂肪组织(ewat)(图20c)和腹股沟白色脂肪组织(iwat)(图20d)的重量。示出了平均值
±
sem,student未配对t检验,与对照相比,*p《0.05。与hfd饲喂的对照相比,hfd饲喂的lko小鼠具有较低的肌肉质量和较多的脂肪组织。
35.图21a和图21b。图21a示出了肝样品(hfd)中指示基因的相对mrna表达,其相对于rpl13a mrna表达进行归一化。来自hfd饲喂的lko小鼠的肝脏中调节脂肪形成的关键基因升高。图21b提供了显示肝样品中指示蛋白质表达的代表性蛋白质印迹。来自lko(hfd)小鼠的肝脏中调节脂肪形成的蛋白质升高。示出了平均值
±
sem;student未配对t检验,与对照相比,*p《0.05。
36.图22a至图22d示出了来自图21b中的蛋白质印迹的指示蛋白质的表达的光密度分析:图22a(fasn);图22b(pparγ);图22c(cd36);图22d(pampk/总ampk)。
37.图23a和图23b。图23a是显示肝甘油三酯(tg)合成途径的示意图;与对照(ctrl)肝脏相比,粗体分子在hfd lko中上调。图23b示出了lko肝脏样品中通过rt-qpcr的指示基因的相对mrna表达,其相对于rpl13a mrna表达进行归一化。lko(hfd)肝脏中调节甘油三酯合成的基因升高。
38.图24示出了来自图21b中的蛋白质印迹的gyk表达的光密度分析。
39.图25a和图25b。图25a是比较hfd饲喂的ctrl和lko小鼠中通过质谱法(lc-ms)测量的肝脂质的火山图。lko(hfd)肝脏中高度升高的脂质种类显示在图的右上象限。图25b示出了维持rd的ctrl和lko小鼠的肝脏中通过rt-qpcr的指示基因的相对mrna表达,其相对于rpl13a mrna表达进行归一化。示出了平均值
±
sem,student未配对t检验,与对照相比,*p《0.05。
40.图26a至图26i。图26a和图26b示出了在12小时禁食后,hfd饲喂的ctrl和lko小鼠中葡萄糖耐量试验(gtt)期间的空腹血糖水平(图26a)和血糖水平(图26b)。与同样hfd饲喂的对照相比,hfd饲喂的lko小鼠的2型糖尿病恶化。图26c示出了gtt的曲线下面积(auc)。图26d示出了在胰岛素耐量试验(itt)期间禁食6小时后,hfd饲喂的ctrl和lko小鼠的血糖水平。图26e示出了itt的auc。ctrl和lko(hfd)肝脏样品中通过rt-qpcr的指示基因的相对mrna表达,显示了调节葡萄糖代谢(图26f)、炎症(图26g)和纤维化(图26h)标志物以及线粒体氧化应激(图26i)的基因,其相对于rpl13a mrna表达进行归一化。来自hfd饲喂的lko小
鼠的肝脏显示出调节炎症和纤维化的基因水平升高。示出了平均值
±
sem,student未配对t检验,与对照相比,*p《0.05。
41.图27是显示kiss1r的相对mrna表达的图。
42.图28a和图28b是显示通过kiss1r信号传导调节肝ppar-γ的蛋白质印迹。lko小鼠肝脏具有增加的ppar-γ表达但降低的磷酸化ppar-γ表达(图28a)。该磷酸化位点抑制ppar-γ活性。相比之下,tak-448治疗降低了ppar-γ的表达但增加了磷酸化ppar-γ,表明ppar-γ活性降低(图28b)。
43.图29a是分离的原代肝细胞中的内源吻素免疫染色(见箭头)的共焦图像。
44.图29b是显示tak-448(3nm)或吻素-10(100nm)对原代小鼠肝细胞中负载ffa(150μm油酸和150μm棕榈酸)的甘油三酯积累的抑制作用的图。
45.图29c是显示tak-448或吻素-10对分离自lko小鼠(缺乏kiss1r)的肝细胞中负载ffa(150μm油酸和150μm棕榈酸)的甘油三酯积累没有影响的图。*p《0.05;单向方差分析,然后是dunnett事后检验。
46.图30是显示tak-448治疗对调节从头脂肪生成和甘油三酯合成的基因表达的抑制作用的图。
47.图31a至图31c。图31a示出了在基础条件下吻素(kp)或tak处理8小时后,分离自c57bl6雄性小鼠的原代小鼠肝细胞中通过rt-qpcr的指示基因的相对mrna表达。(n=5)*p《0.05;单向方差分析,然后是dunnett事后检验。图31b示出了代表性蛋白质印迹,显示了原代小鼠肝细胞(n=4)中,吻素处理对ampk磷酸化(导致其活化)的影响,从而抑制其下游底物acc。图31c示出了通过rt-qpcr的来自ctrl和lko小鼠的原代小鼠肝细胞中的kiss1r表达,其相对于rpl13a mrna表达进行归一化(n=4)。在来自lko小鼠肝脏的分离的肝细胞中kiss1r表达被耗尽。
48.图32a和图32b提供了图31b的蛋白质印迹的光密度分析。
49.图33a和图33b。图33a提供了代表性的蛋白质印迹,显示了在来自对照和lko小鼠的分离的原代肝细胞中指示蛋白质的表达。图33b示出了分离自ctrl和lko小鼠(n=4)的原代小鼠肝细胞中通过rt-qpcr的指示基因的相对mrna表达,显示在kiss1r缺失后调节脂肪合成的基因增加。示出了平均值
±
sem,student未配对t检验,*p《0.05。
50.图34a至图34d提供了图33a的蛋白质印迹的光密度分析,针对cd36(图34a)、fas(图34b)、pparγ(图34c)和mogat1(图34d)。示出了平均值
±
sem,student未配对t检验,与对照相比,*p《0.05。
51.图35a至图35b是一组图,显示负载游离脂肪酸(ffa:棕榈酸酯,100μm)的小鼠原代肝细胞中的tak-448处理增加耗氧率,导致增强的脂肪酸氧化(图35a)。tak-448处理还增加基础呼吸(图35b)和atp产生(图35c)。
52.图36a至图36c是显示负载ffa(棕榈酸酯,100μm)的人肝细胞中的tak-448处理在处理后随时间增加耗氧率的图。tak-448处理还增加基础呼吸(如图36b所示)和(图36c)中所示的atp产生。
53.图37a和图37b是一组条形图,显示tak-448处理减少人星状lx2细胞的增殖,如通过细胞计数减少所观察到的(图37a)。tak-448处理降低了lx2细胞中的纤维化标志物基因表达(图37b)。
54.图38a至图38d是一组免疫荧光图像,示出了来自nafld患者的人肝活检的的染色,如指示对于细胞核(图38a)、内源kiss1r(图38b)、星状细胞(图38c)和叠加(图38d)。
55.图39a是显示患者肝活检中的相对mrna表达的图。与健康对照肝脏相比,来自nafld/nash患者的肝活检中kiss1和kiss1r的基因和蛋白质表达升高。
56.图39b是一组蛋白质印迹;
57.图39c和图39d示出了图39b中印迹的光密度分析。
58.图39e是一组代表性组织学图像,显示与健康受试者相比,来自nafld/nash患者的成人肝脏中的kiss1r染色更高。
59.图39f是通过放射免疫测定显示人类受试者中血浆吻素水平的条形图;与健康受试者相比,nafld/nash患者的血浆吻素水平显著升高,这表明吻素的增加可能是一种旨在解决nafld的补偿性保护机制。
60.图40是显示肝脏中的kp/kiss1r激活防止脂肪肝发展及其进展为nash的信号传导通路的工作模型的示意图。通过有效的类似物tak-448增强kiss1r的激活导致ampk的激活,从而抑制脂肪生成并减少脂肪积累。相反,tak-448通过β-氧化促进脂肪酸分解以产生能量,例如在线粒体中。tak-448还抑制ppar-γ的表达和活性,ppar-γ是肥胖条件下肝脏中甘油三酯合成的关键促进因子。这导致脂肪积累更少,肝细胞损伤(炎症)更少,从而抑制进展为nash。
具体实施方式
61.1.定义
62.除非另有定义,否则本文使用的所有技术和科学术语具有与本领域普通技术人员通常理解的相同含义。尽管在本发明的实践或测试中可以使用与本文所述的那些相似或等效的各种方法和材料,但下文描述了合适的方法和材料。然而,本领域技术人员理解所使用和描述的方法和材料是示例并且可能不是适用于本发明的唯一方法和材料。此外,由于测量受固有可变性的影响,因此本文给出的任何温度、重量、体积、时间间隔、ph、盐度、摩尔浓度或重量摩尔浓度、范围、浓度和任何其他测量值、数量或数值表达式旨在为近似值而不是精确或关键数字,除非有明确相反的说明。
63.如本文所用,术语“约”是指记载值加或减20%,例如,“约0.125”是指0.125
±
0.025,“约1.0”是指1.0
±
0.2。
64.如本文所用,术语“tak-448”是指吻素-54的完全活性10-氨基酸c末端(吻素-10)的稳定类似物;这些是由kiss1基因编码的吻素-145的肽产物。吻素-54以前称为转移抑制素(metastin),是以前称为gpr54的g蛋白偶联吻素1受体(kiss1r)的配体。
65.如本文所用,术语“治疗(treat)”及其同源词诸如“治疗(treatment)”是指获得所需的药理学和/或生理学效果。因此,“治疗”包括(a)防止病症或疾病或其症状在可能易感于该病症或疾病但尚未被诊断为患有它们的受试者中发生;(b)抑制病症或疾病或其症状,诸如阻止其发展;以及(c)减轻、缓解或改善病症或疾病或其症状,诸如,例如,引起病症或疾病或其症状的消退或部分消退。
66.如本文所用,术语“预防(prevent)”及其同源词诸如“预防(prevention)”是指降低疾病或病症发生的可能性,降低疾病或病症的严重性,以及部分或完全停止受试者中的
疾病或病症。
67.如本文所用,术语“受试者”是指哺乳动物,优选人类患者,包括人类儿童患者。
68.如本文所用,术语“有此需要”是指患有或怀疑患有非酒精性脂肪肝病(nafld)和非酒精性脂肪性肝炎的受试者。
69.如本文所用,术语“治疗有效量”是指产生治疗效果、优选地期望的药理学和/或生理学结果的量、剂量或剂量方案。
70.术语“给药(administer)”及其同源词诸如“给药(administration)”是指根据药学领域已知的方法和途径使化合物与受试者接触。
71.术语“脂肪肝”是指其中受试者的肝脏包含5%或更多脂肪的病症。属于“脂肪肝”类别的病症包括但不限于脂肪肝病、肝炎、非酒精性脂肪肝病、非酒精性脂肪性肝炎、酒精性脂肪性肝炎、肝纤维化、肝硬化、黄疸和肝癌症,或肝脏包含5%或更多脂肪的任何病症。
72.如本文所用,术语“nafld/nash”是指非酒精性脂肪肝病和/或非酒精性脂肪性肝炎。nafld包括一系列病理病症,包括脂肪变性(脂肪肝)、非酒精性脂肪性肝炎(nash)、纤维化和肝硬化(肝脏的过度瘢痕化)。
73.就天然或合成肽或肽类似物而言,术语“保守变体”是指与肽约90%或更多相同的序列,例如,在序列中替换、添加或缺失一个氨基酸。该术语还包括肽的化学变体,诸如氨基酸侧链的化学修饰,包括将甲基、乙基、卤素、三氟、羟基、羧酸根/酯、氨基、甲基氨基等添加到化合物的结构中。
74.2.概述
75.本发明通过给药tak-448提供了对代谢紊乱诸如nafld和nash的治疗,tak-448是血液中发现的天然存在的肽吻素-10的长效合成类似物。吻素通过g蛋白偶联受体吻素受体(kiss1r)进行信号传导,并且吻素和kiss1r均由肝脏表达。吻素-54肽已给药于健康男性,已被证明可增强胰岛素分泌,而不会改变食物摄入或食欲。
76.3.本发明的实施方式
77.该报告提供了kiss1r作为肝脂肪生成的关键调节子的新功能的第一个证据。虽然kiss1r在肝脏中表达,但是其在肝脏中的生物学功能仍然未知。本研究的目的是确定kiss1r在脂肪肝诸如nafld的发展和进展中的作用,并提供用于预防和治疗nafld/nash和其他涉及肝脏中脂肪积累的病症的组合物和方法。使用肝脏kiss1r敲除(lko),我们发现肝脏kiss1r缺乏会加剧nafld小鼠模型中的肝脏脂肪变性、炎症和纤维化。hfd lko小鼠表现出加重的代谢参数,诸如增加的体重、升高的肝脏甘油三酯(tg)水平、升高的空腹血糖和胰岛素抵抗以及炎症和纤维化标志物增加。
78.这些表型表明肝脏kiss1/kiss1r消融在负调节nafld表型的发展和相关代谢恶化中起关键作用。因此,为了检验肝脏kiss1r在nafld中起保护作用的假设,用tak-448治疗胰岛素抵抗的野生型小鼠,tak-448是具有与kp10相当的有效激动剂活性的蛋白酶抵抗的kp类似物。我们发现,在胰岛素抵抗的野生型小鼠中,tak治疗抑制肝脏中的脂质积累,降低了血清tg、胆固醇和游离脂肪酸(ffa)并降低了脂肪量。此外,tak治疗在不改变食物摄取的情况下防止体重增加,并改善葡萄糖耐量和胰岛素敏感性。重要的是,tak给药导致调节炎症和纤维化的关键基因降低。总的来说,这些发现证明了kiss1r对脂肪肝(脂肪变性)和nash发展的关键保护作用。人类数据显示,与健康受试者相比,来自nafld/nash患者的肝活检中
kiss1和kiss1r水平显著增加,并且血浆吻素水平升高。这可能是一种降低肝脏脂肪积累的保护性补偿机制。然而,kiss1r的内源激活显然不足以防止nafld的发展。这些结果说明了我们临床前研究结果的转化相关性,因为它们反映了在饮食诱导的nafld小鼠模型中观察到的结果。重要的是,tak-448治疗还减少了在肝纤维化中起关键作用的人肝星状细胞的增殖,表明tak-448在预防纤维化中具有保护作用。
79.我们还描绘了kiss1r调节脂肪生成的细胞通路(参见图40)。我们的研究结果表明,kiss1r在体内激活hfd肝脏中并在体外激活分离的原代肝细胞中的ampk,导致抑制从头脂肪生成(dnl)和tg积累,背后是kiss/kiss1r对脂肪变性的保护作用。dnl(碳水化合物向脂肪的转化)在nafld中上调,并且ampk激活通过使acc磷酸化并降低其活性来抑制dnl,促进脂肪酸的利用。tak-448治疗通过称为β-氧化的过程增加了游离脂肪酸的分解。ampk激活还通过直接使脂肪生成的主要转录调节子srebp-1磷酸化导致脂肪生成基因表达的下调。
80.已显示ampk激活减弱tg合成,通过两种可能的机制产生抗脂肪变性作用。首先,ampk激活抑制肝脏中的肝脏x受体α(lxrα)活性;lxrα是一种脂质传感器,其促进脂肪酸合成并导致高甘油三酯血症。其次,ampk激活抑制pparγ转录。kp激活ampk的机制目前尚不清楚。ampk的激活需要苏氨酸172(t172)的磷酸化,其通过增加amp:atp比率和提高细胞内ca2+(38)实现。几份报告表明,ca2+/钙调蛋白依赖性蛋白激酶β(camkkβ)的激活在哺乳动物细胞中激活ampk中起生理作用。由于kiss1r激活增加了细胞内ca2+(41),因此kiss1r很可能通过camkkβ途径激活ampk。
81.pparγ表达在健康肝脏中低,但在nafld中显著升高,并通过调节从头脂质代谢促进肝脏脂肪变性的发展。此外,pparγ在直接上调参与tg合成的基因/途径中起主要作用,诸如脂肪酸酯化中的关键酶gyk1、脂肪输入因子cd36和将单酰基甘油酯化形成tg前体甘油二酯(dag)的mogat1。pparγ依赖的mogat1表达的抑制会抑制肝脂肪变性,而cd36依赖的ffa摄取和mogat介导的脂肪酸酯化促进脂肪变性。在患有nafld的人中也观察到pparγ和mogat过表达。考虑到dag负调节肝脏胰岛素敏感性之间已建立的联系,tak抑制pparγ、cd36和mogat1表达的发现具有重要的临床意义。总之,我们的研究表明,肝脏kp/kiss1r信号传导系统通过涉及ampk-pparγ信号传导的机制抑制肝细胞中的肝脏从头脂肪生成,进一步表明kiss1r信号传导的激活是用于治疗nafld的有希望的治疗靶标。
82.具体地,本文所述的临床前研究使用已建立的nafld小鼠模型,其中与正常饮食(rd,4%kcal脂肪)相比,向雄性小鼠饲喂西方高脂肪饮食(hfd,60%kcal脂肪)。使用该模型,我们发现与用载剂(磷酸盐缓冲盐水,pbs)对照治疗的小鼠相比,在小鼠nafld模型中皮下给药吻素类似物(tak-448)可防止脂肪肝的发展(参见图1a至图1j以及图3a和图3c)。具体地,将tak-448给药于糖尿病小鼠1个月(图2a和图2b)可防止脂肪变性(肝脏甘油三酯的积累)并防止肝脏和血清甘油三酯升高(图1c),并降低血液胆固醇(图3b)、脂肪酸(图1d)和作为甘油三酯结构单元的甘油(图3c)。tak-448治疗降低了肝酶alt的水平,alt是脂肪肝病的临床生物标志物。见图3a。tak-448治疗还可以防止外周脂肪积累和胰岛素抵抗。见图if至图1j。与对照组相比,tak治疗的小鼠的食物摄取没有变化,两者均饲喂hfd(图2c)。
83.此外,在hfd饲喂的具有肝脏kiss1r缺失(lko)的小鼠中,观察到与hfd饲喂的同窝崽对照相比体重(图14)、肝脏脂肪变性(图15)和肝脏甘油三酯(图16)增加更多,尽管食物摄取没有变化(图14d)。这暗示了吻素/kiss1r通路在预防nafld发病机制中的保护作用。参
见图14、图15和图16。与对照相比,kiss1r敲除动物也表现出葡萄糖不耐受和胰岛素抵抗(全部饲喂hfd)。参见图26b至图26e。与对照相比,来自肝脏kiss1r敲除小鼠的肝脏显示出促炎细胞因子(白细胞介素(il)-lα、mip2、ip10;参见图26g)和nash/纤维化的生化标志物(例如tgfβ、胶原蛋白(参见图26h)显著增加(全部饲喂hfd)。
84.吻素还对脑具有作用,调节中枢生殖系统。长期给药吻素导致性类固醇激素合成的抑制。长效tak-448模仿了这一点,其已用于患有前列腺癌的男性(两项1期临床试验)以降低睾酮水平。此外,另一个名称的tak-448(mvt-602)正在由myovant进行测试治疗女性不孕症,并且目前处于1期和2期临床试验中。
85.tak-448是吻素-54的完全活性10-氨基酸c末端(吻素-10)的寡肽类似物。其在血液中的半衰期为4小时,相比之下,吻素会在几分钟内迅速降解。tak-448是有效的吻素1受体(kiss1r,或gpr54)激动剂。kiss1r是与吻素结合的g蛋白偶联受体。吻素由转移抑制基因kiss1编码,该基因在多种内分泌和性腺组织中表达。吻素和kiss1r在哺乳动物生殖中发挥关键作用,因为它们参与青春期的开始和对下丘脑-垂体-性腺轴的控制。它已被用于研究治疗前列腺癌、低睾酮、前列腺肿瘤和促性腺激素性腺功能减退症的试验。tak-448的结构如下:
[0086][0087]
tak-448
[0088]
感兴趣的序列参见下表1。
[0089]
表1.肽序列
[0090][0091]
可以根据本发明的实施方式治疗的受试者包括任何哺乳动物,包括实验室动物、伴侣动物、农场动物、动物园动物等,包括人。优选的受试者是人和啮齿动物。用于本发明的合适受试者优选是怀疑患有、已被诊断患有或具有风险患上可通过给药tak-448改善、治疗或预防的疾病(包括涉及脂肪肝或肝脏中的脂肪积累的任何疾病或病症)的人。
[0092]
可以给药tak-448以预防、延迟、减缓、逆转涉及脂肪在肝脏中积累或脂肪肝的任何疾病或病症或停止其疾病进展。这样的病症包括但不限于(1)nafld、(2)nash、(3)脂肪肝病、(4)肝炎、(5)肝纤维化、(6)肝硬化、(7)黄疸、(8)肝癌、或(9)受试者的肝脏包含5%或更多脂肪的任何病症。
[0093]
如本文所用,术语“nafld/nash”是指非酒精性脂肪肝病和/或非酒精性脂肪性肝炎。nafld包括一系列病理病症,包括脂肪变性(脂肪肝)、非酒精性脂肪性肝炎(nash)、纤维化和肝硬化(肝脏的过度瘢痕化)。
[0094]
预期用于本文描述的本发明的化合物包括但不限于tak-448或其任何保守变体,以及吻素-10或其任何保守变体。保守变体是具有约90%或更多序列同一性的肽,或如上所述的化学修饰肽。例如,吻素-10的保守变体包括吻素-10的缺失、置换和添加,以及化学变体,诸如例如下表2至表6中举例说明的那些。tak-448的保守变体包括tak-448的缺失、置换和添加,以及化学变体,诸如例如下表2至表6中举例说明的那些。
[0095]
表2.示例性化合物变体。
[0096][0097]
表3.吻素-10类似物的序列和结合亲和力
[0098]
肽名称序列ic50****,nmseq id no吻素-10ynwnsfglrf-nh21.0+/-0.318ana1*anwnsfglrf-nh223.2+/-14.319ana2*yawnsfglrf-nh2145.1+/-138.020ana3*ynansfglrf-nh22.7+/-0.721ana4*ynwasfglrf-nh243.5+/-15.522ana5*ynwnafglrf-nh20.8+/-0.423ana6*ynwnsaglrf-nh2109.5+/-89.824ana7*ynwnsfalrf-nh24.6+/-1.525ana8*ynwnsfgarf-nh277.4+/-10.026ana9*ynwnsfglaf-nh242.3+/-6.727ana10*ynwnsfglra-nh2
‑‑‑
28ana11*fnwnsfglrf-nh23.5+/-2.129ana12*ydwnsfglrf-nh229.1+/-13.530ana13*ynwdsfglrf-nh2140.2+/-60.631ana14*ynwntfglrf-nh22.3+/-1.632ana15*ynwnsyglrf-nh287.5+/-30.733ana16*ynwnsfgirf-nh239.9+/-8.134ana17*ynwnsfglkf-nh29.8+/-2.635ana18*ynwnsfglrh-nh2378.4+/-266.036ana19*[dy]nwnsfglrf-nh23.6+/-0.337ana20*ynwns[df]glrf-nh2252.9+/-144.038ana21*ynwnsfglr[df]-nh2447.2+/-173.039tak-683ac-dydwntfazaglr(me)w-nh2‑‑‑
40化合物2**ac-ynwnsfglry-nh2‑‑‑
41化合物3**ac-yk(palm-γ-d)w gψ[tz]l ry-nh2‑‑‑
42化合物4**ac-ynk(palm-γ-d)nsfgψ[tz]lry-nh2‑‑‑
43
化合物5**palm-γ-dynwnsfgψ[tz]lry-nh2‑‑‑
44化合物6**palm-γ-dynwnsfgψ[tzjlr[me]y-nh2‑‑‑
45化合物7**ac-ynwnsfgψ[tz]lr[me]y-nh2‑‑‑
46
[0099]
*来自curtis et al.,2009的信息;**来自decourt et al.,2016的信息;****ic
50
(平均值+/-se;n=3-5)针对[
125
i]kp-54结合cho-kiss1r膜制剂中的kiss1r。每个受体结合测定一式三份进行。
[0100][0101]
表4.吻素类似物,asami et al.(在第50和51位之间置换的转移抑制素类似物的生物活性。
[0102][0103]
a在人ot7t175的双功能测定中评估所有肽的激动剂活性,这是一种使用荧光成像读板技术的细胞内钙动员测定。使用s型剂量反应曲线计算所有肽类似物的ec
50
值。
[0104]
b转移抑制素(45-54)的ec
50
值计算为13次独立实验的平均值。
[0105]
c合成肽的受体结合亲和力测定为k50值。
[0106]
d转移抑制素(45-54)的ic
50
值计算为三次独立实验的平均值。
[0107]
e在37℃下在小鼠血清中温育1小时后的残留率。
[0108]
f未测定。
[0109]
表5.在第47位置换的吻素类似物,asami et al.(在第47位置换的十肽转移抑制素类似物的生物活性)。
[0110][0111]
a在人ot7t175的双功能测定中评估所有肽的激动剂活性,这是一种使用荧光成像读板技术的细胞内钙动员测定。使用s型剂量反应曲线计算所有肽类似物的ec
50
值。
[0112]
b转移抑制素(45-54)的ec
50
值计算为13次独立实验的平均值。
[0113]
c合成肽的受体结合亲和力测定为k50值。
[0114]
d转移抑制素(45-54)的ic
50
值计算为三次独立实验的平均值。
[0115]
e在37℃下在小鼠血清中温育1小时后的残留率。
[0116]
f未测定。
[0117]
表6.吻素类似物,nishizawa et al.(2019)kp类似物的结构、生物活性和hplc保留时间。
[0118]
[0119]
[0120][0121]
a在表达人kiss1r的cho细胞中评估了所有肽类似物的[ca2+]增加活性的ec50值[nm(95%置信区间)]。
[0122]
b肽类似物的保留时间(tret)通过rp-hplc测量。洗脱条件:merckperformance rp-100mm上的线性密度梯度洗脱,洗脱液a/b=95/5-35/65(10min),使用0.1%tfa水溶液作为洗脱液a和包含0.1%tfa的乙腈作为洗脱液b;流速:3.0ml/min。
[0123]
本发明的化合物还包括碱和任何药学上可接受的水合物、溶剂化物、酸或盐,并且可以是无定形的或任何结晶形式,或作为油或蜡。如果方便,可以使用任何药学上可接受的盐。
[0124]
通常,这些盐衍生自药学上和生物学上可接受的无机或有机酸和碱或金属。这样的盐的实例包括但不限于:乙酸盐、己二酸盐、藻酸盐、铵盐、天冬氨酸盐、苯甲酸盐、苯磺酸盐(苯磺酸盐(besylate))、碳酸氢盐、硫酸氢盐、丁酸盐、柠檬酸盐、樟脑酸盐、樟脑磺酸盐、碳酸盐、环戊烷丙酸盐、二葡萄糖酸盐、十二烷基硫酸盐、乙磺酸盐、甲酸盐、富马酸盐、葡庚酸盐、甘油磷酸盐、乙醇酸盐、半硫酸盐、庚酸盐、己酸盐、盐酸盐、氢溴酸盐、氢碘酸盐、2-羟基乙磺酸盐、乳酸、镁、马来酸盐、丙二酸盐、甲磺酸盐(mesylate)、2-萘磺酸盐、烟酸盐、硝酸盐、草酸盐、棕榈酸盐、果胶酸盐、过硫酸盐、3-苯基丙酸盐、磷酸盐、苦味酸盐、新戊酸盐、钾、丙酸盐、水杨酸盐、钠、琥珀酸盐、硫酸盐、酒石酸盐、硫氰酸盐、甲苯磺酸盐(tosylate)和十一酸盐。
[0125]
化合物还包括治疗剂的任何或所有立体化学形式(即,每个不对称中心的r和/或s构型)。因此,治疗剂的单一对映异构体、外消旋混合物和非对映异构体都在本发明的范围内。治疗剂的空间异构体和位置异构体也在本发明的范围内。一些实施方式的治疗剂还意在包括仅在一个或多个同位素富集原子的存在方面不同的化合物。例如,其中一个或多个原子被例如氘、氚、
13
c、
14
c(或本领域常用的任何同位素标记,诸如磷、钙、碘、氯、溴或用于同位素标记的任何其他方便的元素)也在本发明的范围内。
[0126]
诸如tak-448的化合物和根据本发明实施方式的其他化合物可以通过本领域已知
的任何合适的途径或方法给药于受试者。合适的给药途径包括但不限于口服给药,静脉内、动脉内、鞘内、腹膜内、皮内、皮下、肌肉内或腹膜内注射,通过栓剂或灌肠剂的直肠或阴道给药,经粘膜、经皮、颊、鼻、吸入、舌下、外用或局部给药到靶组织中或上,或通过将治疗有效量的药物或组合物递送至其靶向的细胞或组织的任何途径或方法给药。当口服给药时,优选的剂型是延迟释放或允许化合物在肽或肽类似物化合物降解之前被吸收的其他制剂。优选地,本发明实施方式的方法涉及皮下或静脉内给药。给药可以通过输液或输注进行,并且可以通过植入物、植入泵或外部泵或本领域已知的任何装置给药。
[0127]
在优选的方法实施方式中,本文所述的化合物被配制并作为药物组合物给药,所述药物组合物包括药学上可接受的载体和一种或多种剂,包括一种或多种本文所述的本发明化合物,并且包括任选地具有用于联合治疗的另外的剂(诸如相同或另一类的脂肪肝减少药物)的一种或多种本文所述的本发明化合物。
[0128]
用于与本发明的化合物诸如tak-488同时或依次进行联合治疗的示例性化合物包括但不限于acc抑制剂、ampk、akr-001、aramchol、asc40、azd7687、bi-1467335、bio89-100、bms-986036、卡格列净、cenicriviroc、昔洛法克索、达格列净、dgatedp-305、elafibranor、elobixibat、恩格列净(empagliflozin)、恩利卡生、艾塞那肽(exenatide)、eyp001a、falcon1(peg-fgf21(fasn抑制剂)、非诺贝特、非索考司他(fxr激动剂)、gr-md-02、ionis-dgat2
rx
、伊格列净、利可格列净、利拉鲁肽(liraglutide)、鲁格列净、fy3202328、met409、mge-3196、mk-4074、msdc-0602k、mt-3995、ngm282、尼度法克索、oat-1251、奥贝胆酸(obeticholic acid)、oca、owe833、pf-05221304、pf-06835919、pf-06865571、pf-06882961、吡格列酮(pioglitazone)、px-102、pxe770、pxs-5153a、seladelpar、司隆色替、semaglutide、辛妥珠单抗、tern-101、tern-201、troifexor、ttp273、维生素d、维生素e、vk2809和volixibat。此外,通过给药化合物tak-448、吻素及其变体进行的治疗可以伴随一线治疗,包括饮食和运动改变以及体重减轻。
[0129]
药学上可接受的载体是指无毒并且不会破坏或显著降低与其一起配制的治疗剂的药理学活性的任何方便的化合物或化合物组。这样的药学上可接受的载体或载剂包括本领域已知的任何标准药学上可接受的固体、液体或气体载体,例如本领域中讨论的那些。合适的载体取决于预期用于药物组合物的给药途径。
[0130]
预期用于本发明的载体和载剂的非包含性列表如下:填充剂、稀释剂、佐剂、ph调节剂、缓冲剂、防腐剂、粘合剂和崩解剂、溶剂、脂质、脂质体、乳液、悬浮液和容器(例如,安瓿、瓶子、预装注射器等)。
[0131]
液体载体可以是溶液、悬浮液、乳液、油、凝胶等形式,并且包括例如水溶液(例如,盐溶液、磷酸盐缓冲盐溶液、林格液等)、水包油或油包水乳液、脂质体等。气体载体可以包括例如空气、氧气、碳氟化合物、分散剂等。固体载体可以包括例如淀粉(例如玉米淀粉、马铃薯淀粉、大米淀粉等)、纤维素(例如微晶纤维素等)、糖类(例如乳糖、蔗糖、葡萄糖等)、粘土、矿物质(例如滑石等)、树胶、调味剂、防腐剂、着色剂、掩味剂、甜味剂、脂质、油、溶剂、盐溶液、乳化剂、助悬剂、润湿剂、分散剂、粘合剂、润滑剂(例如硬脂酸镁等)、盐、ph调节剂(例如酸或碱)、缓冲剂等。
[0132]
适用于注射的药物组合物包括无菌水溶液(在水溶性的情况下)或分散体、悬浮液或乳液,以及用于临时制备无菌可注射溶液或分散体的无菌粉末或颗粒。对于静脉内给药,
合适的载体包括生理盐水、抑菌水、cremophor el
tm
(basf
tm
,parsippany,n.j.)或(例如磷酸盐)缓冲盐水(pbs)。在所有情况下,组合物必须是无菌的并且应该是流体以达到易于注射的程度。其在制造和储存条件下应该是稳定的,并且必须被保存以防止微生物诸如细菌和真菌的污染作用。载体可以是溶剂或分散介质,其包含例如水、乙醇、多元醇(例如甘油、丙二醇和液体聚乙二醇等),及其合适的混合物。可以保持适当的流动性,例如通过使用包衣诸如卵磷脂,在分散体的情况下通过保持选定的粒度以及通过使用表面活性剂。可以通过各种抗菌剂和抗真菌剂来防止微生物的作用,例如对羟基苯甲酸酯、氯丁醇、苯酚、抗坏血酸、硫柳汞等。在一些情况下,组合物中包含等渗剂,例如糖、多元醇诸如甘露醇、山梨糖醇、或氯化钠。可通过在组合物中包含延迟吸收的剂,例如单硬脂酸铝或明胶来实现可注射组合物的延长吸收。
[0133]
延长和持续释放组合物也被考虑用于或用在本发明的实施方式。因此,合适的载体可以包括任何已知的成分以实现活性成分的延迟释放、延长释放或持续释放。
[0134]
给药途径由技术人员根据方便性、待治疗受试者的健康和状况以及待治疗病症的位置和阶段来确定。因此,药物组合物可以采取的形式将包括但不限于片剂、胶囊、囊片、锭剂、糖衣丸、丸剂、颗粒剂、口服溶液、用于稀释的粉末、用于吸入的粉末、蒸气、气体、用于注射或输注的无菌溶液或其他液体、透皮贴剂、口腔贴剂、插入物和植入物、直肠栓剂、阴道栓剂、乳膏、洗剂、油、软膏、局部覆盖物(例如伤口覆盖物和绷带)、悬浮液、乳剂、脂质囊泡等。
[0135]
治疗方案包括单次给药或持续两天或更长时间的给药疗程,包括一周、两周、数周、一个月、两个月、数月、一年或更长时间,包括在受试者的余生中给药。方案可以包括例如每天多个剂量、每天或每周一个剂量,或持续一小时、数小时、一整天或更长时间的长时间输注给药。
[0136]
合适的剂量可以由治疗医师根据患者、待治疗的病症和待治疗病症的严重程度来确定。这样的剂量可包括产生所需结果的任何量、剂量或剂量方案。每次给药的剂量包括由医师确定的任何量,并且将取决于待治疗的受试者的体型、受试者的健康状况、给药途径、待治疗或预防的病症等。
[0137]
一般而言,预期对于大多数受试者,约0.01nmol/小时至约10nmol/小时范围内的剂量是合适的,优选地约0.1nmol/小时至约5nmol/小时是可用的,其在约1小时至约72小时、或约24小时至约48小时、或几天、几周或更长的时间段内给予。该剂量可以每周、每天或每天多次给药,或作为连续输注、透皮制剂或贮库制剂给药。
[0138]
此外,可以给药约0.01mg、约0.02mg、约0.025mg、约0.05mg、约0.1mg、约0.2mg、约0.5mg、约1mg、约2mg、约3mg、约4mg、约5mg或更多的单次注射剂量。替代地,可以给药约0.002μmol/kg、约0.004μmol/kg、约0.008μmol/kg、约0.01μmol/kg、约0.025μmol/kg、约0.5μmol/kg、约0.75μmol/kg、约1μmol/kg、约2μmol/kg、约5μmol/kg、约8μmol/kg、约10μmol/kg或更多的单次注射剂量。
[0139]
4.结果总结
[0140]
与hfd饲喂的同窝崽对照相比,尽管食物摄取没有变化,但用hfd(西方高脂肪饮食,60%kcal脂肪)饲喂的肝脏吻素受体(kiss1r)敲除小鼠具有更多的体重增加、肝脏脂肪变性、升高的肝脏甘油三酯以及更高水平的血液甘油三酯和alt(脂肪肝的临床标志物)。这暗示了吻素/kiss1r通路在nafld和其他脂肪肝病症的发病机制中的病理作用。
[0141]
与对照相比,肝脏kiss1r敲除小鼠还是葡萄糖不耐受和胰岛素抵抗的(全部饲喂hfd)。与对照相比,来自肝脏kiss1r敲除小鼠肝脏的肝脏表现出nash/纤维化生化标志物(例如tgfβ、胶原蛋白)和促炎细胞因子(白细胞介素(il)-lα、mip2、ip10)显著增加(全部饲喂hfd)。
[0142]
与用载剂(pbs)对照处理的小鼠相比,皮下给药tak-448防止hfd饲喂小鼠的nafld(脂肪肝或脂肪变性)发展及其进展为nash。
[0143]
向hfd饲喂的小鼠给药tak-448一个月防止肝脏甘油三酯的积累以及血液中甘油三酯、游离脂肪酸和甘油(甘油三酯结构单元)的升高。与hfd饲喂的载剂处理的对照(其发生了nafld)相比,这些变化在食物摄取没有变化的情况下发生。
[0144]
tak-448治疗还可以防止外周脂肪积累和胰岛素抵抗,并通过减少调节炎症和纤维化(nash的标志)的关键基因的表达来防止nash的发展。
[0145]
5.实施例
[0146]
本发明不限于所描述的特定过程、组合物或方法,因为它们可以变化。描述中使用的术语仅用于描述特定版本或实施方式的目的,并不旨在限制本发明的范围,本发明的范围将仅由所附权利要求限制。尽管与本文所述的那些相似或等效的任何方法和材料可以用于本发明的实施方式的实践或测试,但是现在描述优选的方法、装置和材料。本文提及的所有出版物均通过援引整体并入;本文中的任何内容均不得解释为承认本发明无权因在先发明而早于此类公开。
[0147]
实施例1:方法和材料。
[0148]
a.动物。
[0149]
动物研究根据鲁特格斯大学机构动物护理和使用委员会制定的指导方针获得批准。通过使雄性alb-cre+/-与雌性kiss1rrf/fl杂交来产生kisslr alb-cre敲除小鼠。alb-cre小鼠购自jackson laboratories
tm
。kiss1rrf/fl小鼠根据已知方法产生。f1代是kiss1rfl/+,albcre+/-或kiss1rfl/+,albcre-/-。使kiss1rfl/+,albcre+/-与kiss1rfl/fl回交以产生kiss1rfl/flalb,albcre+/-(肝脏敲除:lko)和kiss1rf l/fl(同窝崽对照)。将小鼠安置在保持12小时光/暗循环的无病原体屏障设施中。
[0150]
给4周大的雄性lko和对照小鼠饲喂高脂肪饮食(hfd:60%kcal脂肪、0.2796%胆固醇、20%来自碳水化合物的卡路里(research diets
tm
目录#d12492,new brunswick,nj)或普通对照食物(rd)。将小鼠分组安置并随意提供食物和水。开始指定饮食后五个月,进行葡萄糖耐量试验,一周后进行胰岛素耐量试验。在开始指定饮食后每周测量总体重,持续20周,然后对动物实施安乐死。
[0151]
b.kp-类似物tak-448的给药。
[0152]
将c57bl6/j雄性小鼠(4周龄)维持hfd或rd 6周,然后测量空腹血糖。根据已知方法,将包含tak-448(0.3nmol/小时)或pbs(载剂对照)的azlet
tm
微型渗透泵模型2004皮下插入小鼠的侧腹。除非另有说明,否则对动物进行4周的治疗并维持rd或hfd(共10周)。
[0153]
c.代谢测试。
[0154]
使用血糖仪(bayer contour
tm
)通过侧尾静脉中的小切口获得血糖测量值。对于葡萄糖耐量试验,将小鼠禁食12小时,然后腹腔内注射d-葡萄糖(1g/kg)。对于胰岛素耐量试验,将小鼠禁食6小时,然后腹膜内注射胰岛素(0.5u/kg;novo nordisk
tm
)。
[0155]
d.代谢笼评估。
[0156]
将小鼠单独安置在具有受控光照和进食的8室临床实验动物监测系统(clams)中。在4天的时期内测量二氧化碳输出、氧气更新、呼吸交换率(rer)、走动和进食。
[0157]
e.胰岛素、甘油、甘油三酯、alt、ffa和胆固醇测量。
[0158]
使用超灵敏小鼠胰岛素elisa试剂盒(crystal chem
tm
)测量血清胰岛素水平。使用甘油测定试剂盒(sigma
tm
)测量血清甘油水平。使用甘油三酯定量试剂盒(mbl
tm
国际目录号jm-k622-500)测量肝脏甘油三酯。使用游离脂肪酸定量试剂盒(sigma
tm
,mak044-1kt)测量游离脂肪酸。根据制造商的说明使用液体alt(sgpt)试剂组测量alt水平;根据制造商的说明使用胆固醇组测量胆固醇水平。
[0159]
f.免疫印迹测定。
[0160]
免疫印迹测定根据已知方法进行。将小鼠肝组织或原代肝细胞在包含蛋白酶抑制剂的ripa裂解缓冲液中均质化,并在4℃下离心;通过蛋白质印迹分析来分析裂解物中的蛋白质表达。使用sds-page分离蛋白质并使用以下抗体进行探测:abcam
tm
(兔抗kiss1r 1:1000;兔抗mogat1 1:1000)、cell signaling technology
tm
(兔抗pampk、兔抗ampk、兔抗pacc、兔抗fasn、兔抗ppar-γ、兔抗cd36和兔抗gyk,均为1:1000)、proteintech
tm
(兔抗kiss1 1:750)。小鼠抗β肌动蛋白(1:5000,thermo fisher scientific
tm
)或抗黏着斑蛋白(1:1000,bio-rad
tm
)用于上样对照。然后将蛋白质在辣根过氧化物酶(hrp)缀合的兔(1:2500,cell signaling technology
tm
)或小鼠(1:2500,cell signaling technology
tm
)二抗中温育1小时。使用chemidoc
tm touch成像系统(bio-rad
tm
)、supersignal和west dura extended duration substrate(thermo scientific
tm
)通过化学发光对印迹进行成像。使用image lab software(bio-rad
tm
)对蛋白质水平进行量化。
[0161]
g.定量实时pcr(qpcr)。
[0162]
使用trizol方法从细胞中提取总rna。根据制造商的说明使用iscript rt supermix(bio-rad
tm
)进行逆转录。使用下表7中所示的引物,如先前所述使用sybr green实时qpcr(rt-qpcr)确定基因表达。以下引物购自biorad
tm
(经过验证的pcrprime
tm
引物):srebp1、aqp3、aqp9、gpat2、agpat2、dgat1、dgat2、mogat1、slc2a2、dgkg、dgkh、lgpat1、sod1、sod2、gss、ucp2和gpam。il-13购自qiagen
tm
(qt00099554)。用于srebf1、aqp3、aqp9、gpat2、agpat2、dgat1、dgat2、mogat1、slc2a2、lgpat1、sod1、sod2、gss、ucp2和gpam的引物是预先制备的并且购自biorad
tm
。
[0163]
表7.引物序列。
[0164][0165][0166]
h.组织学分析。
[0167]
在对小鼠实施安乐死后,立即将肝组织固定在10%中性福尔马林中。组织由鲁特格斯大学的研究病理学服务部处理用于组织学。将肝脏切成5μm厚的切片并用苏木精和伊红染色。通过光学显微镜检查切片的组织病理学变化。
[0168]
i.通过液相色谱-质谱法(lc-ms)的代谢组学分析。
[0169]
根据已知方法,通过lc-ms对血清和肝脏进行代谢组学分析。细胞代谢物的lc-ms分析在q exactive plus hybrid quadrupole orbitrap质谱仪(thermo fisher scientific
tm
)上使用亲水相互作用色谱进行。使用maven软件用每种标记同位素获得数据(质量精度窗口:5ppm)。使用r中编码的accucor包校正标记同位素天然丰度和杂质。
[0170]
j.小鼠原代肝细胞研究。
[0171]
根据已知方法分离来自小鼠的原代肝细胞。使用氯胺酮/甲苯噻嗪混合物(henry schein
tm
)麻醉小鼠(野生型,c57bl6)或lko和同窝崽对照。将肝脏通过肝门静脉插管,并首先用包含egta的37℃克雷布林格溶液灌注10分钟。第一次洗涤后,使用包含cacl2和liberase(roche
tm
)的第二克雷布林格溶液,直到肝脏完全灌注。肝细胞通过纱布网过滤并重悬于具有10%fbs(sigma
tm
)、200nm地塞米松(sigma
tm
)、5mm青霉素/链霉素(fisher
tm
)、2mm l-谷氨酰胺(fisher
tm
)的威廉姆斯培养基e(sigma
tm
)中。将细胞以3x105的密度铺板在6孔胶原蛋白包被板(sigma
tm
)上。使肝细胞恢复过夜,然后在实验前血清饥饿3小时。对于游离脂肪酸(ffa)加载,使150μm油酸和150μm棕榈酸与2%bsa(0.3mm)缀合。细胞在分离后加载ffa,并在血清饥饿前过夜恢复,然后用kp-10或tak处理。
[0172]
k.人肝细胞heparg细胞
[0173]
人肝heparg细胞购自thermo fisher scientific
tm
。细胞在补充有10%胎牛血清、青霉素-链霉素(10,000u/ml)、2mm l-谷氨酰胺、5微克/ml胰岛素(humulin r.)和50微摩尔氢化可的松半琥珀酸盐的威廉姆斯e培养基中生长至汇合。通过向生长培养基中补充2%dmso两周来使汇合细胞分化,以获得包含肝细胞样和肝祖细胞二者的汇合分化培养物,然后将细胞传代并用于实验。
[0174]
l.甘油三酯合成
[0175]
使游离脂肪酸(150μm油酸和150μm棕榈酸)与2%bsa(0.3mm)缀合。细胞在分离后加载ffa,并在血清饥饿前过夜恢复,然后用kp-10(r&d systems
tm
)或tak-448(medchemexpress
tm
)处理。使用甘油三酯定量试剂盒(mbl
tm
国际目录号jm-k622-500)测量tg。
[0176]
m.免疫荧光
[0177]
对于免疫荧光研究,将细胞如所述固定在福尔马林中,用triton
tm x透化,并染色。细胞与kiss1抗体(emd millipore
tm
,1:250)然后与山羊抗兔af555(invitrogen
tm
;1:400)一起温育。细胞核用hoechst
tm
(invitrogen
tm
;1:10000)染色。使用zeiss
tm lsm 700激光扫描显微镜获取图像。
[0178]
n.患者血液采集和血浆吻素测量
[0179]
该研究已获得鲁特格斯大学机构审查委员会的批准,并且所有研究参与者(男性)都提供了书面同意。排除患有可能影响葡萄糖或脂质代谢的慢性疾病的个体,包括活动性恶性肿瘤、hiv感染、乙型肝炎、丙型肝炎、酗酒、慢性胰腺炎、活动性病毒/细菌感染、严重心力衰竭或呼吸衰竭。测定了健康受试者和患有脂肪肝(nafld,n=16)或nash(n=8)的患者
的血浆kp免疫反应性。nafld诊断是基于在没有其他肝病原因的情况下ast和alt水平升高以及超声显示存在肝脂肪变性。nash诊断基于揭示大泡性脂肪变性、小叶和门静脉炎症和纤维化的组织学分析。简而言之,将来自新泽西州新不伦瑞克的罗伯特伍德约翰逊医学院(robert wood johnson medical school in new brunswick,nj)的亚专科诊所招募的受试者的血液(5ml)收集在bd vacutainer k2 edta管(vwr international)中。血液以1210xg离心10分钟,并且将血浆立即冷冻保存。使用商业吻素elisa测量血浆kp免疫反应性。
[0180]
o.免疫组织化学
[0181]
小鼠肝脏由鲁特格斯大学的研究病理学服务部处理用于组织学。对冷冻肝组织的低温切片进行油红o染色以检测中性脂质。健康人肝脏的石蜡切片购自origene technologies
tm
,inc.(md,usa)。来自nash/nafld患者的石蜡切片是在病理学家确认诊断后,从存放在罗伯特伍德约翰逊大学医院病理学实验室的存档肝组织中获得的。脱蜡和热诱导抗原修复后,将载玻片与兔多克隆抗gpr54(abeam
tm
,ab137483;1:1000)温育,然后是immpress
tm hrp抗兔igg(过氧化物酶)聚合物检测试剂盒(vector laboratories
tm
)。
[0182]
p.耗氧率(ocr)
[0183]
根据制造商的方案,使用分离的原代小鼠肝细胞和人肝heparg细胞,使用agilent seahorse biosciences
tm
细胞外通量分析仪(xfe24)测量ocr。简而言之,将细胞在500μl威廉姆斯e培养基(10%fbs、200nm地塞米松、1%青霉素链霉素和2mm谷氨酰胺)中以每孔2
×
104个细胞接种在xf24板中,并与100μm bsa-pal或bsa+/-tak-448(3nm)在37℃和5%co2下温育过夜。在测定前将细胞血清饥饿1小时。在具有0.5mm葡萄糖和0.5mm l-肉碱的seahorse
tm xf培养基中,用tak-448(3nm)或pbs处理,进行基础ocr测量。使用wave软件(agilent
tm technologies)分析结果。
[0184]
q:统计分析。
[0185]
通过未配对双尾student t检验确定两组之间的统计学显著性。对于多组之间的比较,使用单因素方差分析然后dunnett多重比较检验。p值《0.05被认为是统计学显著的。图是使用graphpad
tm prism 8.3.1版(san diego,ca)生成的。
[0186]
实施例2:初步研究:在高脂肪饮食诱导的nafld的临床前小鼠模型中,吻素类似物(tak-4481)给药预防nafld(脂肪肝及其进展为nash)并防止胰岛素抵抗的发生。
[0187]
将c57bl/6j小鼠用高脂肪饮食(hfd)或对照正常饮食(rd)饲喂12周(4-5只/组)。使用皮下植入的alzet微型渗透泵给药tak-448(tak)或载剂(veh,即pbs对照)4周。结果见图1。脂肪变性由苏木精和伊红染色的小鼠肝脏(脂质积聚、白色小液滴、箭头)和由对照肝脏中脂质的油红o染色(灰点)显示(图1a)。这在tak-448治疗组中被阻止(图1a)。图ib至图1g分别显示终点肝甘油三酯(tg)、终点血液tg、血液游离脂肪酸、血液甘油、体重、附睾白色脂肪组织脂肪积累和腹股沟白色脂肪组织脂肪积累均通过tak-448给药而降低。tak-448治疗防止空腹血糖升高(肝糖异生的测量)并防止葡萄糖耐受不良和胰岛素抵抗,如分别通过葡萄糖耐量试验(gtt)或胰岛素耐量试验(itt)测量的。见图1h至图1j(黑色圆圈)对比于对照(空心圆圈)。*,p《0.05;student t检验或单向方差分析和dunnett事后检验。可给药tak-448以在(1)nafld患者(有或没有ii型糖尿病)和(2)nash患者中延缓或防止疾病进展。
[0188]
实施例3:tak-448治疗不会改变nafld小鼠模型中的食物摄取。
[0189]
为了确定增强的kiss1r信号传导对nafld发展的影响,将野生型c57bl6j小鼠(4周龄)置于rd或hfd下6周。hfd饲喂的野生型小鼠体重增加(图2a)并产生胰岛素抵抗,导致在tak-448之前空腹血糖水平升高(糖尿病;图2b)。然后向小鼠(同窝崽,具有类似体重)输注载剂(pbs)或kp类似物(tak,0.3nmol/小时),并在rd/hfd下再维持4周。如预期的,hfd对照(veh)小鼠与rd(veh)对照小鼠相比具有显著更高的体重(图1e)。然而,在hfd小鼠中,tak治疗的小鼠体重明显低于veh对照,并且空腹血糖水平低于veh组对照(图1e);这些变化在食物摄取没有变化的情况下发生(图2c)。与这些表型一致,与hfd对照相比,hfd tak治疗的小鼠葡萄糖耐受(图1i)和胰岛素敏感(图1j)。
[0190]
实施例4:在高脂肪饮食(hfd)诱导的nafld的临床前小鼠模型中,吻素类似物(tak-448)治疗降低了肝酶(alt)以及循环甘油三酯和游离脂肪酸水平,并减少了白色脂肪组织质量。
[0191]
除了其对肝脏甘油三酯积累和葡萄糖稳态的显著影响外,在hfd饲喂的小鼠中的tak-448治疗还可以防止脂肪肝病的临床标志物alt升高(参见图3a)并减少血液胆固醇(参见图3b)和甘油(参见图3c)。clams分析进一步揭示,经治疗和未经治疗的小鼠之间的能量产生或呼吸交换率(rer)没有差异(图4a和图4b)。
[0192]
机制上,hfd条件下的tak治疗显著降低了tg合成和脂肪合成的关键调节子诸如pparγ、cd36和mogat1的肝脏表达(图5a;图5b;图6a;图6b;图6c)。此外,tak治疗诱导肝脏中ampk的磷酸化(图7;图8a),其在激活后抑制pparγ转录。ampk激活还通过急性抑制3-磷酸甘油酰基转移酶(gpat)活性和通过负调节srebp1转录来抑制脂质合成。在tak治疗的肝脏中gpam(编码gpat)表达显著降低(图5a),这可能导致随后的gpat活性降低。尽管srebf1的降低并不显著,但其下游靶标fasn显著降低(图5b;图8b)。tak治疗降低了脂肪生成的重要调节子lfabp1和gyk1的表达(图5a)。
[0193]
最后,tak治疗的肝脏具有较低水平的促炎基因(ip10、mcp1;图9a),以及降低水平的纤维化标志物(cola2、acta2;图9b)和氧化应激标志物(ucp2、gss;图9b)。总体而言,数据表明,体内tak治疗通过激活ampk下调脂质合成来防止脂肪变性,并且这会减弱nafld的发展。
[0194]
acc的磷酸化降低其活性,从而抑制从头脂肪生成(参见图31b;图32b)。从rd肝脏kiss1r敲除(lko)小鼠分离的肝细胞表现出cd36、fas、pparγ和mogat1水平升高(参见图33a、图33b;图34a至图34d)。总的来说,基于数据,我们提出kiss1r的激活通过激活ampk来负调节肝脏脂质含量,其然后抑制从头脂肪生成,此外还抑制pparγ(甘油三酯合成的关键调节子)及其下游信号通路(见图40)。
[0195]
图40是更新的工作模型,显示了kiss1r激活抑制向nash进展的信号通路(参见虚线箭头)。吻素(kp)类似物(tak-448)激活kiss1r会激活主要能量传感器(ampk),使其磷酸化。激活的ampk然后通过使其下游靶酶acc磷酸化而导致其抑制从而抑制脂肪生成,来抑制脂肪生成(脂肪的形成)。这也导致线粒体中促进脂肪酸的β氧化的关键酶cpt1的激活,从而导致脂肪分解。此外,ampk的激活通过降低调节tg合成途径中许多酶表达的转录因子ppar-γ的活性和表达来抑制甘油三酯(tg)合成。总之,这导致肝脏中脂肪积累的减少,从而抑制nafld及其进展为nash和纤维化。
[0196]
实施例5:tak-448给药抑制肝脏甘油三酯合成和从头脂肪生成的关键调节子的表
达。
[0197]
图10呈现了与通过qpcr分析的指示基因(甘油三酯合成和脂肪形成(脂肪生成)的调节子)的表达有关的数据。对比于对照,*p《0.05;student未配对t检验。这些数据表明,tak-448治疗降低了肝脏甘油三酯(tg)合成和从头脂肪生成的关键调节子的表达。对于每个基因,表达(是什么基因,它们参与什么)都显著降低,因此tak-448治疗可用于降低患者肝脏中的脂肪。
[0198]
实施例6:tak-448给药抑制肝脏炎症的关键调节子的表达。
[0199]
通过pcr分析检查了炎症调节中的指定关键基因(巨噬细胞炎症蛋白(mip2)、趋化因子、干扰素γ诱导蛋白10(ip10)、白细胞介素1α(il1a)、促炎细胞因子肿瘤坏死因子(tnf)-α(tnfa)、单核细胞趋化蛋白(mcp1)和白细胞介素1α(il1a)和1β(il1b))的表达,并且结果呈现在图11中。对比于对照,*p《0.05;student未配对t检验。结果表明,tak-448治疗降低了这些肝脏炎症的关键调节子的表达。
[0200]
实施例7:tak-448给药抑制肝纤维化的关键调节子的表达。
[0201]
通过qpcr分析的调节纤维化的指示基因(胶原蛋白(colla2)、平滑肌肌动蛋白(acta2)和转化生长因子β(tgfb))的表达显示在图12中。对比于对照,*p《0.05;student未配对t检验。结果表明,这种吻素类似物可降低肝纤维化关键调节子的表达。
[0202]
实施例8:tak-448给药提高调节脂肪的线粒体和过氧化物酶体β-氧化的基因的表达。
[0203]
tak-448治疗提高了线粒体脂肪酸转运的限速酶cpt1a和cpt2的表达,并且还提高了调节脂肪酸过氧化物酶体β-氧化的限速步骤的酰基辅酶a氧化酶(aox)的表达。对比于对照,*p《0.05;student未配对t检验。参见图13,其显示了通过qpcr分析的指示基因的表达。总之,tak-448(吻素类似物)治疗增加了调节脂肪的线粒体和过氧化物酶体β-氧化的肝脏基因的表达,这将导致脂肪分解增加。
[0204]
实施例9:kiss1r的肝脏敲除(ko)促进nafld小鼠模型中的体重增加、肝脏脂肪变性和脂质积累。
[0205]
通过使kiss1r fl/fl小鼠与alb-cre小鼠(jackson labs
tm
)杂交产生肝脏kiss1r ko(lko)。向同窝崽对照(kiss1r fl/fl)和kiss1r ko小鼠(3周大,10只/组)饲喂正常饮食(rd)或高脂肪饮食(hfd)20周。尽管食物摄取没有变化(图14),但lko小鼠的体重显著增加(图14a至图14c)。h&e染色的小鼠肝脏显示出lko肝脏中脂肪变性与对照相比显著增加(均饲喂hfd)(图15a)。相比之下,在维持rd的动物的肝脏中没有观察到脂肪变性(参见图15b,其显示了健康的肝脏)。方框区域在下部图像中被放大(图15b)。图16显示了与同样hfd饲喂的对照(ctrl)相比,lko(hfd)小鼠中的肝脏甘油三酯升高。*,p《0.05student t检验或单向方差分析和dunnett事后检验。
[0206]
实施例10:产生肝脏kiss1r敲除以确定肝脏kiss1r在调节nafld中的作用。
[0207]
a.肝脏kiss1r缺乏加剧了肥胖、胰岛素抵抗小鼠中的肝脂肪变性。
[0208]
为了测试肝脏kiss1/kiss1r是否参与nafld的发病机制,在饮食诱导的nafld小鼠模型中测量肝脏kiss1/kiss1r表达。在向4周大的野生型c57bl/6小鼠饲喂高脂肪饮食(hfd)8周后,肝脏中的kiss1和kiss1r mrna水平显著增加。参见图17,其显示了饲喂正常饮食(rd)或高脂肪饮食(hfd)8周的c57bl6雄性小鼠中通过rt-qpcr的kiss1和kiss1r的相对
rna表达,其相对于rpl13a mrna表达进行归一化。因此,高脂肪饮食(hfd)诱导吻素(kiss1)和吻素受体(kiss1r)基因的表达。
[0209]
接下来,为了研究kiss1r在调节肝脏脂质代谢中的作用,产生了kiss1r的肝脏特异性敲除(lko)。对lko小鼠的分析表明kiss1r表达显著降低,但kiss1没有。参见图18a。然后将lko小鼠及其同窝崽对照置于对照正常饮食(rd)或hfd下。饲喂rd的lko和对照小鼠的体重没有差异(图18b)。在组之间没有观察到rer(呼吸交换率)和走动活动的差异(参见图18c和图18d)。
[0210]
有趣的是,与对照相比,hfd饲喂的lko小鼠表现出较低的热量消耗(指示较低的代谢)(图19a;通过clams评估的热量消耗)。与rd相比,两个hfd组的血清丙氨酸转氨酶(alt)水平(脂肪肝病的临床标志物)均升高,指示hfd诱导的肝细胞损伤(图19b)。
[0211]
与hfd饲喂的对照相比,lko小鼠还表现出肌肉质量减少(图20a(腓肠肌)和图20b(胫骨前肌)),以及腹股沟白色脂肪组织增加,尽管在附睾白色脂肪组织中没有观察到变化(参见图20c和图20d)。lko hfd小鼠肝脏tg的增加表明肝脏kiss1r在肝脏中对脂肪变性起保护作用。
[0212]
b.肝脏kiss1r缺乏上调调节脂肪生成和游离脂肪酸(ffa)摄取的基因。
[0213]
为了阐明lko小鼠中肝脏脂质积累的潜在机制,测量了脂肪酸摄取的肝脏调节子(脂肪酸转位酶分化簇(cd36)、肝脏脂肪酸结合蛋白(lfabp1))以及脂肪生成(过氧化物酶体增殖物激活受体γ(pparγ,由pparg编码)、甾醇调节元件结合蛋白-1(srebp1,由srebf1编码)及其下游靶标脂肪酸合酶(fas,由fasn编码))的水平。在hfd条件下,调节脂肪形成的关键基因显著增加(图21a;肝脏样品(hfd)中指示基因的相对mrna表达,其相对于rpl13a mrna表达进行归一化),除了acaca,其编码乙酰-coa羧化酶1(acc1,催化从头脂肪酸合成的第一个关键步骤)增加但不显著。与对照相比,hfd lko肝脏中ppar-γ及其下游基因靶标cd36的蛋白质水平以及fas水平也显著升高(图21a;图22a、图22b和图22c))。示出了平均值
±
sem;student未配对t检验,与对照组相比,*p《0.05。
[0214]
此外,lko肝脏表现出ampk磷酸化的抑制(图21b;图22d),ampk是在激活时通过负调节srebf1及其下游基因靶标acaca和fasn而抑制从头脂肪生成的蛋白激酶。
[0215]
实施例11:肝脏kiss1r敲除表现出增加的调节甘油三酯合成的基因的表达和提高的肝脂质水平。
[0216]
甘油三酯(tg)合成需要3-磷酸甘油(g3p),其可以通过两种方法形成:(a)甘油激酶(gyk)依赖的甘油磷酸化和(b)磷酸二羟丙酮的3-磷酸甘油脱氢酶(gpd1)依赖的还原。参见图23a,其示出了肝甘油三酯(tg)合成途径的示意图;粗体分子在hfd lko和对照(ctrl)肝脏中上调。对来自hfd lko小鼠的肝脏的分析揭示了gyk1 mrna和蛋白质水平的肝脏表达显著增加(图23b、图21b和图24)。hfd饲喂的肝脏kiss1r敲除(lko)小鼠表现出调节甘油三酯合成的基因水平升高(参见图23b)。
[0217]
甘油主要通过水甘油通道蛋白(aqp)诸如aqp3和aqp9进入肝脏(参见图23a)。在lko hfd小鼠肝脏中aqp9 mrna水平显著上调,而aqp3水平保持不变(图23b)。在lko hfd肝脏中,许多调节tg合成的酶也上调,包括催化tg合成中的限速步骤的gpat1(由gpam编码)、将dag乙酰化形成tg的二酰基甘油(dag)酰基转移酶2(dgat2)以及将单酰基甘油转化为tg的直接前体二酰基甘油(参见图23b)的单酰基甘油酰基转移酶1(mogat1)(参见图23a,显示
hfd饲喂的lko小鼠表现出调节甘油三酯合成的基因水平升高)。总之,这表明lko小鼠表现出升高的调节tg合成的基因的水平。
[0218]
为了揭示导致在hfd条件下的lko小鼠中观察到的不同表型的代谢差异,对lko和对照肝脏进行了全局、非靶向代谢组学分析。这揭示了包括tg、dag和磷脂酰胆碱在内的多种脂质在hfd lko肝脏中显著上调(参见图25a,通过质谱法显示在hfd饲喂的lko小鼠中肝脏脂质代谢物增加的火山图)。在患有nafld和nash的患者中也观察到了类似的观察结果。lko小鼠还表现出其他变化。这些包括高水平的神经酰胺、磷脂酰甘油和心磷脂。据报道,神经酰胺合成的抑制会减轻肝脂肪变性和纤维化,而线粒体磷脂磷脂酰甘油与包括肝脂肪变性在内的多种代谢疾病有关。心磷脂是一种对最佳线粒体功能至关重要的磷脂,其改变会导致多种组织中的线粒体功能障碍,包括胰岛素抵抗和nafld。
[0219]
由于数据显示肝脏kiss1r的丢失导致hfd下的肝细胞中脂质积累的增加,其可能是通过pparγ及其下游基因靶标的上调,我们询问这是否也发生在维持rd的lko小鼠的肝脏中。有趣的是,维持rd的lko小鼠没有发生脂肪变性或积累tg。然而,pparg显著上调,而mogat1、cd36或srebf1没有显著变化(参见图25b)。这表明kiss1r信号传导调节肝脏脂肪生成的潜在机制是通过调节pparγ。
[0220]
实施例12:肝kiss1r ko小鼠是葡萄糖不耐受的和胰岛素抵抗的。
[0221]
由于胰岛素抵抗在nafld的发病机制中起重要作用,因此进行了代谢测试以检查肝脏kiss1r损失对血糖水平的影响。结果显示,与hfd对照相比,hfd lko小鼠具有显著更高的空腹血糖水平,指示升高的糖异生(参见图26a)。它们也是葡萄糖不耐受的(图26b和图26c)和胰岛素抵抗的(图26d和图26e)。
[0222]
此外,在lko hfd肝脏中,调节糖异生的关键肝基因的表达增加,诸如g6pc(在糖异生的最后一步将葡萄糖-6-磷酸转化为葡萄糖)、pck1(将草酰乙酸转化为磷酸烯醇式丙酮酸)和由slc2a2编码的肝葡萄糖转运蛋白glut2(图26f)。
[0223]
nafld可进展为nash,这是一种与肝脏中增加的炎症、纤维化和氧化应激相关的状态。我们观察到在lko小鼠中,hfd 20周后,调节与nafld相关的炎症的多种基因上调,诸如细胞因子巨噬细胞炎症蛋白2(mip2)、白细胞介素1亚型illα和ilβ(由il1a和illb编码)和趋化因子干扰素γ诱导蛋白10(ip10)以及单核细胞趋化蛋白(mcp1)。参见图26g。
[0224]
lko小鼠还表现出参与nash的基因增加,其在纤维化的早期阶段上调。这些包括胶原蛋白(colla2)和转化生长因子β(tgfb1)。还观察到平滑肌肌动蛋白(acta2)和金属蛋白酶组织抑制剂1(timp1)的增加,但这没有达到显著性水平(参见图26h)。此外,氧化应激标志物(sod1、sod2、gss)增加(参见图26i)。总之,这些发现表明肝脏kiss1r信号传导的损失对肝脏产生有害影响,促进了nafld表型。
[0225]
实施例13:kiss1r敲除(lko)在肝脏中是选择性的。
[0226]
hfd饲喂的ctrl和lko小鼠中的各种器官通过rt-qpcr的基因(图27指示)表达,显示了kiss1r敲低的特异性,在肝脏中具有选择性。对比于对照,*p《0.05;student未配对t检验。
[0227]
实施例14:肝kiss1r信号传导负调节肝ppar-γ表达和活性。
[0228]
转录因子ppar-γ是在nafld患者和实验小鼠模型的脂肪性(脂肪)肝中诱导的脂肪生成的关键调节子。图28呈现了代表性的蛋白质印迹,显示了抑制位点(丝氨酸-112)处
ppar-γ的磷酸化降低,尽管来自hfd饲喂的ctrl和lko小鼠的肝脏中ppar-γ的表达升高。参见图28a。相反地,图28b显示与载剂(pbs)处理的对照相比,tak-448的给药降低了小鼠肝脏中的ppar-γ表达并诱导其在丝氨酸-112处的磷酸化,导致ppar-γ靶基因mogat1、cd36和fas的表达降低。参见图5b。
[0229]
实施例15:分离的原代小鼠肝细胞:tak-448处理通过抑制从头脂肪生成和甘油三酯合成的关键调节子的表达直接抑制甘油三酯积累。
[0230]
由于数据显示kiss1r信号传导抑制体内脂肪变性,因此使用原代小鼠肝细胞检查了tak-448对肝脂肪生成的直接影响。图29a呈现了原代肝细胞中内源吻素(kiss1蛋白,见箭头)免疫染色的代表性共焦图像。比例尺,50μm。在存在或不存在与牛血清白蛋白(bsa)载体缀合的游离脂肪酸(ffa:150μm棕榈酸酯和150μm油酸酯)混合物的情况下培养肝细胞。分离自对照小鼠的负载ffa的肝细胞的kp-10(100nm)或tak-448(3nm)处理导致tg积累减少(参见图29b)。然而,图29c显示tak-448和kp-10未能抑制分离自肝kiss1r敲除(lko)小鼠的肝细胞中的甘油三酯积累。tak-448处理(8小时)还降低了在存在或不存在与牛血清白蛋白(bsa)载体缀合的游离脂肪酸(ffa:150μm棕榈酸酯和150μm油酸酯)混合物的情况下培养的分离自c57bl6雄性小鼠的原代小鼠肝细胞中调节从头脂肪生成和甘油三酯(tg)合成的基因的表达(图30)。吻素(kp)或tak处理8小时降低了新生脂肪生成基因的基础表达(图31a)并刺激了ampk及其下游靶标acc的磷酸化(图31b),这表明tak-448处理激活了分离的原代肝细胞中的ampk,从而抑制脂肪合成。图31c显示在分离自lko小鼠的原代肝细胞中kiss1r表达被耗尽。另参见图32,其示出了图31b中代表性蛋白质印迹的光密度分析,显示吻素处理对原代小鼠肝细胞中ampk及其下游底物acc的磷酸化的影响(n=4)。
[0231]
实施例16:分离的原代小鼠肝细胞:分离自kiss1r敲除小鼠的肝细胞中从头脂肪生成和甘油三酯合成的关键调节子的表达增加。
[0232]
分离自kiss1r敲除小鼠的肝细胞表现出调节脂肪形成的蛋白质诸如cd36、fas、pparγ和mogat1的水平升高(参见图33a、图34a至图34d,后者示出了图33a中印迹的光密度分析)。分离自kiss1r敲除小鼠的肝细胞也显示出调节脂肪形成的基因水平升高(图33b)。
[0233]
实施例17:分离的原代小鼠肝细胞:tak-448处理增加游离脂肪酸的线粒体β氧化。
[0234]
图35示出了在具有或没有tak-448(3nm)或cpt1抑制剂乙莫克舍(etomoxir,eto)的情况下用100μm棕榈酸酯(pa)或bsa载剂对照)处理的肝细胞中的耗氧率(ocr);cpt1是线粒体β氧化的关键调节子。在依次用2.5μm寡霉素(oligo)、3μm线粒体氧化磷酸化解偶联剂羰基氰化物-4(三氟甲氧基)苯腙(fccp)、和2.5μm复合体i和iii抑制剂鱼藤酮和抗霉素a(r+a)处理之后,使用seahorse分析仪的代表性ocr迹线如图35a所示。该图显示tak-448增加了线粒体脂肪酸氧化(fao)。图35b显示tak-448增加基础呼吸,并且图35c显示tak-448增加atp产生。示出了平均值
±
sem。student未配对t检验,对比于对照,*p《0.05;单向方差分析,然后dunnet事后检验。结果表明,tak-448(吻素类似物)处理直接增加了游离脂肪酸的线粒体β-氧化,即促进其在分离的原代小鼠肝细胞中的分解。
[0235]
实施例18:人肝细胞:tak-448处理增加游离脂肪酸的线粒体β氧化。
[0236]
图36a呈现了在分别用1μm寡霉素(oligo)、1μm fccp和0.5μm鱼藤酮和抗霉素a(r+a)依次处理后人肝heparg细胞中的耗氧率(ocr)的seahorse
tm
分析仪迹线。在存在或不存在tak-448(3nm)的情况下,用100μm棕榈酸酯(pa)或bsa处理细胞。tak-448处理增加了人肝细
胞中脂肪的分解。图36b显示tak-448增加基础呼吸,并且图36c显示tak-448增加人肝细胞中的atp产生。示出了平均值
±
sem。student未配对t检验,或单向方差分析,然后dunnett事后检验;对比于对照,*p《0.05。因此,结果表明tak-448(吻素类似物)处理增加了人肝细胞中游离脂肪酸的线粒体β氧化。
[0237]
实施例19:人肝星状lx-2细胞:tak-448处理抑制增殖并减少纤维化标志物。
[0238]
将20万个lx-2细胞接种到每个孔中,并用tak-448(3nm)或载剂(pbs)处理48小时,然后使用血细胞计数器计数。参见图37a中的结果。将细胞用tak-448(3nm)处理48小时。然后,通过rt-qpcr测量纤维化标志物(平滑肌肌动蛋白:acta和胶原蛋白:col1a1)的基因表达变化。student未配对t检验,*p《0.05;n=3。参见图37b中的结果。这些结果表明tak-448抑制人肝星状lx-2细胞增殖和纤维化基因表达。
[0239]
实施例20:人nash活检切片:内源吻素受体定位于肝星状细胞。
[0240]
检查了人nash活检切片中内源吻素受体(kiss1r)和星状细胞标志物(结蛋白(desmin))的定位。使用hoechst
tm
对细胞核进行染色。左侧看到的免疫细胞浸润对于kiss1r染色是阴性的(参见图38a,箭头)。kiss1r与结蛋白(肝星状细胞的分子标志物)共定位的区域显示为亮白色(在叠加中,图38d,箭头)。
[0241]
实施例21:人nafld中kiss1和吻素受体(kiss1r)表达以及血浆吻素水平升高。
[0242]
图39a示出了在患者肝活检中通过rt-qpcr的人kiss1和kiss1r的相对mrna表达,其相对于tbp进行归一化。在nafld/nash患者中kiss1和kiss1r基因表达升高。图39b显示了代表性的蛋白质印迹。示出了平均值
±
sem,student未配对t检验,与对照相比,*p<0.05。图39c和39d示出了来自图39b的蛋白质印迹的光密度分析。图39e示出了使用兔多克隆抗人kiss1r的成人肝脏中内源人kiss1r表达的免疫染色图像的代表性结果;比例尺:80μm。来自nafld/nash患者的患者肝活检中的kiss1r蛋白表达升高。图39f示出了通过放射免疫测定在人类受试者中测量的血浆吻素(kp)水平(pmol/l;平均值
±
sd)。nafld患者肝脏中的吻素(kiss1)蛋白表达升高。下面的表8示出了图39a、39b中男性受试者的临床特征。使用非参数kruskal-wallis检验进行统计分析。误差线:sd。与健康受试者相比,nafld和nash患者中的血浆吻素水平升高。
[0243]
表8.男性受试者临床特征。
[0244]
临床特征健康t2dnafldnash年龄(岁)32.2
±
6.060.4
±
11.9h45.4
±
13.2
h,d
55
±
12.9h体重(kg)74.2
±
9.494.6
±
18.5h97.8
±
22.4h99.5
±
23.3hbmi(kg/m2)23.3
±
4.131.1
±
6.2h31.7
±
5.5h33.6
±
5.8hast(单位/l) 20.7
±
7.538.3
±
55.4d37.4
±
15.3dalt(单位/l) 22.6
±
7.173.6
±
51.9d51.9
±
38.2fib-4评分
ꢀꢀ
0.9
±
06s2.2
±
1.1
[0245]
数据表示为平均值
±
sd。
[0246]
h:与健康相比,p《0.05显著性。
[0247]
d:与t2d相比,p《0.05显著性。
[0248]
s:与nash相比,p《0.05显著性。
[0249]
实施例22:tak-448在nafld中的保护作用机制。
[0250]
这些发现表明,tak-448对kiss1r的增强激活通过激活ampk,然后抑制脂肪生成并增加脂肪酸氧化,此外还抑制调节甘油三酯合成的pparg及其下游信号通路来负调节肝脂质含量。(参见图40)。这导致防止脂肪肝(脂肪变性)的发展及其进展为nash和纤维化。总之,此处提供的数据表明,tak-448是一种有效的吻素类似物,其通过激活ampk和增加线粒体中脂肪的分解(即脂肪酸氧化)来防止脂肪肝的发展及其进展为nash。
[0251]
参考文献
[0252]
下面和整个说明书中列出的所有参考文献均通过援引整体并入本文。
[0253]
1.takeda的美国专利号7,625,869.
[0254]
2.dudek et al.,frontiers endocrinol.,9(145):1-8,2018.
[0255]
3.wang et al.,frontiers physiol.,9(209):1-13,2018.
[0256]
4.song et al.,cell metab.,19(4):667-681,2014.
[0257]
5.tolson et al,j.clin.invest.,124(7):3075-3079,2014.
[0258]
6.wolfe and hussain,frontiers endocrinol.(lausanne),9(184),2018.
[0259]
7.国际申请号pct/jp2007/071169.
[0260]
8.machado cortez-pinto,world j.gastroenterol.20(36):12956-12980,2014.
[0261]
9.younossi et al.,hepatol.,64(1):73-84,2016.
[0262]
10.younossi et al.,hepatol.64(5):1577-1586,2016.
[0263]
11.diehl and day,n.engl.j.med.,377(21):2063-2072,2017.
[0264]
12.wong et al.,gastroenterol.,148(3):547-555,2015.
[0265]
13.bhattacharya and babwah,endocrinol.,156(4):1218-1227.2015.
[0266]
14.seminara et al.,n.engl.j.med.,349(17):1614-1627,2003.
[0267]
15.shoji et al.,peptides,31(10):1920-1925,2010.
[0268]
16.foretz et al.,int.j.mol.sci.,19(9),2018.
[0269]
17.browning and horton,j.clin.invest.,114(2):147-152,2004.
[0270]
18.jelen et al.,j.biol.chem.,286(52):44319-44325,2011.
[0271]
19.rodriguez et al.,cell cycle.,10(10):1548-1556,2011.
[0272]
20.puri et al.,hepatol.,46(4):1081-1090,2007.
[0273]
21.zhang et al.,cell.mol.gastroenterol.hepatol.,7(4):763-781,2019.
[0274]
22.abdelmalek,nat.rev.gastroenterol.hepato.,13(12):685-686,2016.
[0275]
23.diehl and day,n.engl.j.med.,377(21):2063-2072,2017.
[0276]
24.ng et al.,lancet,384(9945):766-781,2014.
[0277]
25.izzi-engbeaya et al.,diabetes obes.metab.,20(12):2800-2810,2018.
[0278]
26.maclean et al.,j.clin.endocrinol.metab.99(8):e1445-e1453,2014.
[0279]
27.asami et al.,j.med.chem.,56(21):8298-8307,2013.
[0280]
28.paradies et al.,antioxid.redox.signal.20(12):1925-1953,2014.
[0281]
29.paradies et al.,world j.gastroenterol.,20(39):14205-14218,2014.
[0282]
30.petrosillo et al.,biochim.biophys.acta,1767(10):1260-1267,2007.
[0283]
31.tarantino et al.,j.clin.med.,9(1),2019.
[0284]
32.friedman et al.,nat.med.,24(7):908-922,2018.
[0285]
33.chang et al.,sci.rep.,5:10096,2015.
[0286]
34.haukeland et al.,j.hepatol.,44(6):1167-1174,2006.
[0287]
35.mirea et al.,trends mol.med.,24(5):458-471,2018.
[0288]
36.syn et al.,clin.liver dis.,13(4):565-580,2009.
[0289]
37.rakhshandehroo et al.,ppar res.2010;2010.
[0290]
38.thompson et al.,am.j.physiol.endocrinol.metab.,291(5):e1074-1082,2006.
[0291]
39.calder et al.,endocrinol.,155(8):3065-3078,2014.
[0292]
40.sozio et al.,am.j.physiol.gastrointest.liver physiol.,301(4):g739-g747,2011.
[0293]
41.li et al.,cell.metab.,13(4):376-388,2011.
[0294]
42.nagarajan et al.,am.j.physiol.endocrinol.metab.,316(4):e578-e589,2019.
[0295]
43.munday et al.,eur.j.biochem.,175(2):331-338,1998.
[0296]
44.millar and babwah,neuroendocrinol.,101(3):193-210,2015.
[0297]
45.lambert et al.,gastroenterol.,146(3):726-735,2014.
[0298]
46.hwahng et al.,hepatol.,49(6):1913-1925,2019.
[0299]
47.smith et al.,am.j.physiol.endocrinol.metab.,311(4):e730-e740,2016.
[0300]
48.green et al.,cell signal.,23(12):2005-2012,2011.
[0301]
49.willows et al.,biochem.j.,474(17):3059-3073,2017.
[0302]
50.kirbyet al.,pharmacol.rev.,62(4):565-578,2010.
[0303]
51.bedoucha et al.,j.hepatol.,35(1):17-23,2001.
[0304]
52.memon et al.,endocrinol.,141(11):4021-4031,2000.
[0305]
53.li et al.,proc.natl.acad.sci.u.s.a.,111(36):13163-13168,2014.
[0306]
54.mazzucotelli et al.,diabetes,56(10):2467-2475,2007.
[0307]
55.lasar et al.,cell.rep.,22(3):760-773,2018.
[0308]
56.guan et al.,genes dev.,19(4):453-461,2005.
[0309]
57.tontonoz et al.,cell,93(2):241-252,1998.
[0310]
58.lee et al.,proc.natl.acad.sci.u.s.a.,109(34):13656-13661,2012.
[0311]
59.yu et al.,sci.rep.,6:29352,2016.
[0312]
60.wolf greenstein et al.,j.endocrinol.,232(1):107-121,2017.
[0313]
61.hall et al.,j.lipid res.,53(5):990-999,2012.
[0314]
62.yu et al.,biochem.biophys.res.commun.,460(3):715-720,2015.
[0315]
63.savage et al.,physiol.rev.,87(2):507-520,2007.
[0316]
64.novaira et al.,mol.endocrinol.,28(2):225-238,2014.
[0317]
65.zajac et al.,plos one,6(6):e21599,2011.
[0318]
66.cvetkovic et al.,endocrinol.,154(6):1999-2014,2013.
[0319]
67.dragan et al.,cell death dis.,11(2):106,2020.
[0320]
68.blake et al.,sci.rep.,7:46525,2017.
[0321]
69.kalemba et al.,j.biol.chem.,294(48):18017-18028,2019.
[0322]
70.curtis et al.,am.j.physiol.endocrinol.metab.298:e296-e303,2009.
[0323]
71.nishizawa et al.,bioorg.med.chem.lett.29(4):654-658,2019.
[0324]
72.decourt et al.,scientific rep.6:26908,2016。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1