HPK1抑制剂及其用途的制作方法
hpk1抑制剂及其用途
1.对相关申请的交叉引用
2.本技术要求2020年7月3日提交的国际专利申请号pct/cn2020/100134的优先权。上述申请的全部内容通过引用并入本文。
3.发明背景
4.造血祖细胞激酶1(hematopoietic progenitor kinase,hpk1),亦称为丝裂原活化蛋白激酶激酶激酶激酶1(mitogen-activated protein kinase kinase kinase kinase 1(map4k1)),其是在典型三层式mapk途径的上游作用的蛋白激酶,该途径包括map3k(map激酶激酶激酶(map kinase kinase kinase)),其激活map2k(map激酶激酶(map kinase kinase)),其进而激活双重thr与tyr mapk家族成员jnk(c-jun n端激酶)。最初在造血祖细胞中克隆的hpk1/map4k1主要在淋巴器官/组织表达,包括骨髓、胎儿肝脏、淋巴结、胎盘、脾脏及胸腺(hu等人,gene&dev.10(18):2251-2264,1996;kiefer等人,the embo j.15(24):7013-7025,1996)。在细胞水平,hpk1在造血区室中的所有细胞类型中表达,包括造血祖细胞、t细胞、b细胞、巨噬细胞、树突细胞、嗜中性粒细胞及肥大细胞(hu,如上述文献,kiefer,如上述文献)。
5.hpk1/map4k1为包括hpk1(map4k1)、gck(map4k2)、glk(map4k3)、hgk/nik(map4k4)、khs/gckr(map4k5)及mink(map4k6)的六种map4k的其中之一。总言之,这些map4k为目前已鉴定出的约26种哺乳动物ste20样丝氨酸/苏氨酸激酶的成员,其是酵母sterile20蛋白质(ste20p)的同源物,ste20p是在酵母信息素信号传导途径中激活map3k的推定map4k。这些哺乳动物ste20样激酶基于域结构分成两个亚家族:p21-活化激酶(pak)及生发中心激酶(gck)。gck亚家族中,有数个成员可激活map3k激酶级联反应,造成jnk活化。
6.map4k具有高度类似的结构,其具有n端激酶域(kd),接着为2-4个富含脯氨酸的基序,及c端枸橼同源(citron-homology)域(cnh)。
7.hpk1的激酶域的atp-结合位点包括lys-46。此残基突变成为met(hpk1-m46)破坏hpk1的催化性活化(hu,如上述文献)。
8.hpk1的激酶域内有多个保守的ser/thr磷酸化位点,并且在其前两个富含脯氨酸的基序之间有保守的tyr磷酸化位点。hpk1激活似乎需要tyr379(小鼠中,或在人中的tyr381)被lck/zap70磷酸化,因为在jurkat t细胞中,受到抗cd3刺激时,lck或zap70的缺陷破坏tyr-379磷酸化及hpk1的激酶活性(ling等人,jbc 276(22):18908-18914,2001;liou等人,immunity 12(4):399-408,2000;sauer等人,jbc 276(48):45207,45216,2001)。另一方面,thr-355自体磷酸化调节hpk1的泛素化与降解。thr-355为靶向pp4的脱磷酸化位点;此脱磷酸化作用防止cul7/fbxw8-介导的泛素化及活化hpk1的蛋白酶体降解(wang等人,cancer res.69(3):1063-1070,2009),因此hpk1亦被蛋白质磷酸酶4(pp4)稳定化及活化(zhou等人,jbc 279(47):49551-49561,2004)。
9.tyr磷酸化位点亦邻接胱天蛋白酶(caspase)切割位点(ddvd)。已显示,全长hpk1可在凋亡细胞中在此位点被胱天蛋白酶-3切割,造成n端hpk1片段的增强的催化活性(chen等人,oncogene 18:7370-7377,1999)。
10.hpk1的四种富含pro的基序介导hpk1与许多含sh3域的蛋白质相互作用(boomer&tan,jcb 95(1):34-44,2005)。
11.hpk1的cnh域可能涉及hpk1-介导的淋巴细胞粘附,因为另一种ste20样激酶tnik的枸橼同源域结合至rap2,并且调节肌动蛋白细胞骨架(taira等人,jbc 279(47):49488-49496,2004)。
12.通过调节细胞信号传导、免疫细胞激活、细胞转化及细胞迁移。hpk1敲除(ko)小鼠显示增强的t-细胞活化,增加细胞因子产生,以及在klh免疫后增加抗体产生,map4k在免疫系统中,特别是在淋巴细胞中起重要作用。hpk1ko小鼠亦更易受eae诱导的影响。hpk1ko t细胞与b细胞显示增强的细胞活化及抗原受体信号传导。hpk1ko树突细胞显示较高的共刺激分子和促炎性细胞因子的水平(alzabin等人,j.immunol.182(10):6187-6194,2009;shui等人,nat.immunol.8(1):84-91,2007)。
13.在细胞系(例如,hek293与cos-1细胞,及造血jurkat t细胞与白血病hl-60细胞)中过表达,证实hpk1可以通过多种map3k(包括tak1、mekk1和mlk3)激活mapk jnk(但不是p38或erk map激酶),其均激活map2k mkk4与mkk7,进而激活jnk。
14.值得注意,map4k在免疫细胞中的调节功能似乎主要受到不依赖jnk的机制介导。已证实ikk-nf-κb活化需要hpk1激酶活化,且认为hpk1是通过调节carma1来运作。carma1为在所谓cbm(carma1/bcl10/malt1)复合物中的转接蛋白,其在jurkat t细胞中受到抗cd3刺激时,促进ikkβ活化。活化的ikk裂解iκb并且释放相关的nf-κb核转录因子。特别地,hpk1可诱导地与carma1相关联,并且直接在ser-551处磷酸化carma1,这是nf-κb活化所必需的(brenner等人,pnas usa,196(34):14508-14513,2009)。
15.t细胞中,当tcr刺激时,淋巴细胞蛋白质酪氨酸激酶(lck)使tcr/cd3复合物的细胞溶质侧的免疫受体酪氨酸活化基序(itam)磷酸化。zap-70随后募集至tcr/cd3复合物,在此进行磷酸化和活化。活化的zap-70使称为slp-76的转接蛋白磷酸化,其转移至质膜,并且通过与包括hpk1的许多蛋白质结合,促进形成多重蛋白质信号小体复合物。这些蛋白质集体传递tcr信号至不同效应分子,造成t-淋巴细胞的活化、存活及增殖。
16.此过程中,hpk1直接结合slp-76的sh2域,并且主要充当tcr信号传导的负调节剂。例如,在hpk1ko原代t细胞中增强tcr信号传导,当在体外连接tcr时,其显示过度增殖及产生il-2(shui,如上述文献)。认为hpk1可通过负反馈机制,在ser-376处磷酸化slp-76转接蛋白,而下调tcr信号传导。当ser-376被hpk1磷酸化时,slp-76通过磷酸化的ser-376残基结合至14-3-3,造成slp-76的lys-30(k30)残基泛素化,随后即成为蛋白酶体降解的靶标。hpk1亦通过类似机制,在其他转接蛋白(包括gads)下调tcr信号传导(例如,通过gads的thr-254的磷酸化及促进14-3-3相互作用)。
17.因此,hpk1似乎在jnk活化及tcr信号传导起双重且相反的作用。虽然hpk1已证实在不同过表达系统中通过map3k-map2k-mapk途径直接激活jnk途径,但hpk1-介导slp-76活化的抑制亦造成tcr信号传导中的jnk活性的抑制。此点与hpk1敲除的原代t细胞显示未受影响的jnk活性结果一致(shui,如上述文献)。同样地,hpk1似乎以两种不同且相对的机制来调节ikk活化-一方面,hpk1通过直接磷酸化carma1来激活ikk;另一方面hpk1也通过抑制slp-76活化来负向调节ikk活化。此点似乎与hpk1所起的双重作用相反,理解hpk1在tcr信号传导初始期促进jnk及ikk活化,但在末期阻碍tcr信号传导中起重要作用。
18.hpk1亦在b细胞中的bcr诱导的细胞活化及增殖中起类似的负向调节作用。b细胞利用称为blnk的类slp-76转接蛋白来转导bcr信号传导,包括jnk及ikk活化。b细胞中,tyr激酶syk及lyn促进hpk1的tyr磷酸化与活化,及造成hpk1的py379介导hpk1-blnk结合作用。blnk的hpk1的负向反馈是通过blnk的thr-152。pt152被14-3-3结合,造成在多重lys残基的blnk泛素化,随后blnk的蛋白酶体降解(因此阻碍bcr信号传导)。
19.值得注意的是hpk1似乎为调节性t细胞(t
reg
)的压制功能的正向调节物(sawasdikosol等人,j immunol.188(supp.1):163,2012)。hpk1缺陷小鼠foxp3
+
tregs在压制tcr诱导的效应子t细胞增殖中有缺陷,并且在tcr接合时,矛盾地得到产生il-2的能力(sawasdikosol,如上述文献)。因此,hpk1为treg功能及外周自体耐受性的重要调节剂。
20.hpk1亦涉及pge2介导的cd4
+
t细胞活化的抑制(ikegami等人,j immunol.166(7):4689-4696,2001)。us2007/0087988显示,cd4
+
t细胞暴露到pge2的生理浓度时,通过pge2诱导的pka活化,可增加hpk1激酶活性。hpk1缺陷的t细胞的增殖可耐受pge2的压制效应(us 2007/0087988)。因此,pge2介导的hpk1活化可能代表调控免疫应答的新调节途径。
21.不同于tcr及bcr,hpk1亦转导tgf-r(转化生长因子受体)(wang等人,jbc 272(36):22771-22775,1997)或gs-偶联pge2受体(ep2及ep4)(ikegami等人,j immunol.166(7):4689-4696,2001)下游的信号。
22.hpk1负向调节免疫细胞粘附。t细胞中,tcr活化亦诱导整合素活化,造成t-细胞粘附及免疫突触形成。此是由促进降解的转接蛋白(adap)的slp-76结合来达成,其是tcr诱导的整合素活化所需的(wang等人,j.exp.med.200(8):1063-1074,2004),但其组成性相关的skap55蛋白质使活化小gtpase rap1靶向质膜,造成整合素活化(kliche等人,mcb 26(19):7130-7144,2006)。换言之,slp-76/adap/skap55三元复合物接力传达tcr信号传导给整合素家族的粘附分子,从而促进t-细胞粘附。hpk1不仅通过下调slp-76(如上述文献),而且通过与adap竞争slp-76上相同的sh2结合位点,而负向调节此途径,进而阻碍adap下游效应子rap1的活性(patzak等人,eur.j.immunol.40(11):3220-3225,2010)。
23.hpk1同样负向调节b细胞中的整合素活化及细胞粘附。其中,hpk1与称为skap-hom的skap55同源物相关(konigsberger等人,plos one 5(9).pii:e12468,2010),其是b-细胞粘附所需(togni等人,mcb 25(18):8052-8063,2005)。认为hpk1诱导skap-hom的负向磷酸化位点,进而压制rap1活化。
24.然而,在嗜中性粒细胞中,hpk1正向调节其粘附。嗜中性粒细胞运送,包括缓慢滚动、紧密结合、细胞扩散及血球渗出,受到β2-整合素活化的由外往内信号传导所控制,诱发肌动蛋白与hip-55(55kda hpk1-相互作用蛋白质)之间的相互作用。其强化β2-整合素的高亲和力构造,有助于嗜中性粒细胞粘附(hepper等人,j.immunol.188(9):4590-4601,2012;schymeinsky等人,blood 114(19):4209-4220,2009)。当β2-整合素介导的粘附作用时,hpk1与hip-55及肌动蛋白共同定位在嗜中性粒细胞的片状伪足(jakob等人,blood 121(20):4184-4194,2013)。cxcl1介导的嗜中性粒细胞粘附作用在体内及体外被hpk1缺陷或hip-55缺陷破坏(jakob,如上述文献schymeinsky,如上述文献)。
25.与hpk1下调tcr及bcr功能的角色一致,hpk1负向调节后天免疫应答,并丧失hpk1介导的调节t-细胞活化,且免疫应答可能为自体免疫发病机制的重要机制。在hpk1ko小鼠中,虽然t及b细胞的发展似乎不受影响(shui,如上述文献),但来自这些动物的t细胞显示,
当在体外受到抗cd3刺激时,tcr近端的信号传导及下游erk的活化大幅增加,造成这些细胞过度增殖(shui,如上述文献)。来自经免疫hpk1-缺陷小鼠的t细胞受到抗原特异性刺激时过度应答,并且产生显著更高水平的促炎性细胞因子,如il-2、ifn-γ及il-4。此类小鼠亦产生更高水平的igm及igg同等型,表示增强hpk1敲除b细胞的功能(shui,如上述文献)。
26.hpk1亦负向控制小鼠的自体免疫性,因为hpk1ko小鼠对诱导实验性自体免疫脑脊髓炎(eae)的敏感性较高(shui,如上述文献)。hpk1衰减亦有助于异常的t-及b-细胞活化及人患者的自体免疫力。银屑病性关节炎患者的外周血单个核细胞或系统性红斑狼疮(sle)患者的t细胞的hpk1已下调。
27.hpk1的生理功能不限于淋巴细胞,hpk1亦通过未知机制负向调节树突状细胞(dc)成熟及活化(alzabin,如上述文献)。在hpk1ko小鼠中,骨髓衍生的树突状细胞(bmdc)显示提高的共刺激分子cd80/cd86水平,以及增加的促炎性细胞因子产生(alzabin,如上述文献)。结果,hpk1ko小鼠中的树突状细胞的抗原呈递活性更高效(alzabin,如上述文献)。更重要的是,由hpk1ko bmdc介导的ctl应答的肿瘤根除比野生型bmdc更有效(alzabin,如上述文献)。此外,hpk1亦可经由t-及b-淋巴细胞依赖性机制控制抗肿瘤免疫性。已显示hpk1缺陷t细胞的过继移转在控制肿瘤生长及转移比野生型t细胞更有效(alzabin等人,cancer immunol immunother 59(3):419-429,2010)。同样地,来自hpk1敲除小鼠的bmdc在提供t细胞应答来根除lewis肺癌(lewis lung carcinoma)比野生型bmdc更高效(alzabin等人,j immunol.182(10):6187-6194,2009)。
28.因此,需要hpk1抑制性化合物通过调控hpk1活性来治疗疾病或病症。
29.发明概述
30.本文描述了抑制hpk1活性的式(i)、(ia)、(ib)、(ii)、(iia)和(iib)的化合物,及其药学上可接受的盐或立体异构体(本文统称为“本发明的化合物”)。
31.本文提供了药物组合物,其包含有效量的本公开的化合物或其药学上可接受的盐或其立体异构体,以及药学上可接受的载剂。
32.还提供了组合,其包含治疗有效量的本公开的化合物或其药学上可接受的盐,和一种或多种治疗活性并用剂(co-agent)。
33.本发明进一步提供了抑制有此需要的受试者中的hpk1活性的方法,该方法包括向该受试者施用治疗有效量的本公开的化合物、其药学上可接受的盐或立体异构体。
34.本公开还提供了治疗患有本文所述疾病或状况,如癌症(如乳腺癌、结直肠癌、肺癌、卵巢癌和胰腺癌)的受试者的方法,该方法包括向该受试者施用治疗有效量的本公开的化合物、其药学上可接受的盐或立体异构体。
35.某些实施方案公开了本公开的化合物、其药学上可接受的盐和立体异构体,其用作药物,如充当hpk1抑制剂的药物。
36.本公开还提供了本公开的化合物、其药学上可接受的盐或立体异构体,或包含其的药物组合物在上述本公开的方法中的任一者中的用途。在一个实施方案中,提供了本公开的化合物、其药学上可接受的盐或立体异构体,或包含其的药物组合物,其用于本文所述本公开的方法中的任一者。在另一个实施方案中,提供了本公开的化合物、其药学上可接受的盐或立体异构体,或包含其的药物组合物在制备用于本公开所述方法中的任一者的药物的用途。
37.发明详述
38.1.概述
39.本文所述的本公开提供了hpk1/map4k1抑制剂、其药学上可接受的盐或立体异构体、其药物组合物、以及使用其调控(例如,抑制)hpk1/map4k1活性的方法,所述方法包括向有此需要的患者/受试者施用本公开的hpk1/map4k1抑制剂化合物,或其药学上可接受的盐。某些实施方案中,本公开的化合物、其药学上可接受的盐或立体异构体用于治疗性施用,以在治疗癌症中增强、刺激和/或增加免疫力。
40.例如,治疗与抑制hpk1相互作用相关联的疾病或病症的方法可以包括向有此需要的患者施用治疗有效量的本文所提供的化合物或其药学上可接受的盐或立体异构体。本公开的化合物可以单独使用、与其他药剂或疗法联合使用,或作为佐剂或新辅助剂(neoadjuvant)使用,用于治疗疾病或病症,包括癌症。
41.2.定义
42.如本文所用,术语“一种”、“一个”、“该”及类似术语用于本发明的上下文中(尤其在权利要求书的上下文中)解释为涵盖单数与复数,除非本文中另有其他说明或明显与上下文矛盾。
43.上述式中的任何一种的化合物可以表现出异构体(例如光学异构体、几何异构体或互变异构体)的一种或多种种类。此类变化是隐含在上述式中的任何一种的化合物中,因为它们是参照其结构特征定义的并且因此在本公开的范围内。
44.具有一个或多个手性中心的化合物可以以各种立体异构形式存在,即每个手性中心可以具有r或s构型,或者可以是两者的混合物。立体异构体是仅在空间排列上不同的化合物。立体异构体包括化合物的所有非对映体和对映体形式。对映体是相互镜像的立体异构体。非对映体是具有两个或更多手性中心的立体异构体,这些手性中心是不相同的并且不互为镜像。
45.实验部分的“峰1”是指从色谱分离/纯化获得的预期反应产物化合物,其洗脱早于来自相同先前反应的第二预期反应产物化合物。第二预期产物化合物被称为“峰2”。
46.当化合物通过其化学名称(例如,在化学名称中以“r”或“s”表示构型)或其结构(例如,以“楔形”键表示构型)指示单一对映体时,除非另有说明,该化合物是至少60%、70%、80%、90%、99%或99.9%光学纯的(也被称为“对映体纯的”)。光学纯度是指混合物中命名或描述的对映体的重量除以混合物中两种对映体的总重量。
47.当公开的化合物的立体化学通过结构命名或描绘,并且命名或描绘的结构涵盖超过一个立体异构体(例如,在非对映对中),应理解为包括所涵盖的立体异构体之一或所涵盖的立体异构体的任何混合物。应进一步理解,命名或描绘的立体异构体的立体异构纯度按重量计为至少60%、70%、80%、90%、99%或99.9%。在这种情况下,立体异构纯度是通过将混合物中名称或结构所涵盖的立体异构体的总重量除以混合物中所有立体异构体的总重量来确定。
48.当两个立体异构体通过其化学名称或结构描绘,并且化学名称或结构由“和”连接时,意指这两个立体异构体的混合物。
49.当两个立体异构体通过其化学名称或结构描绘,并且名称或结构由“或”连接时,意指这两个立体异构体中的一个或另一个,而不是两者。
50.当通过结构描绘所公开的具有手性中心的化合物而不显示该手性中心的构型时,该结构是指涵盖在该手性中心具有s构型的化合物、在该手性中心具有r构型的化合物,或在该手性中心具有r和s构型的混合物的化合物。当通过其化学名称描绘公开的具有手性中心的化合物而没有用“s”或“r”表示该手性中心的构型时,该名称意味着涵盖在该手性中心具有s构型的化合物、在该手性中心具有r构型的化合物或在该手性中心具有r和s构型的混合物的化合物。
51.外消旋混合物是指一个对映体的50%和相应对映体的50%。当命名或描绘具有一个手性中心的化合物而不表明手性中心的立体化学时,理解为该名称或结构涵盖该化合物的两种可能的对映体形式(例如,两种对映体纯的、对映体富集的或外消旋的)。当命名或描绘具有两个或多个手性中心的化合物而不表明手性中心的立体化学时,理解为该名称或结构涵盖该化合物的所有可能的非对映体形式(如非对映体纯的、非对映体富集的和一个或多个非对映体的等摩尔混合物(如外消旋混合物))。
52.术语“几何异构体”是指具有至少一个双键的化合物,其中双键可以以顺式(也称为syn或entgegen(e))或反式(也称为anti或zusammen(z))的形式,及其混合物存在。
53.当通过名称或结构描绘几何异构体,应理解为该命名或描绘的异构体比另一种异构体的存在程度更大,即该命名或描绘的几何异构体的几何异构纯度大于50%,如按重量计算至少60%、70%、80%、90%、99%或99.9%纯。几何异构纯度是通过将混合物中命名或描绘的几何异构体的重量除以混合物中所有几何异构体的总重量来确定。
54.用于制备/分离个别对映体/非对映体的常规技术包括从合适的光学纯前体进行手性合成或使用例如手性高压液相色谱法(hplc)解析外消旋体(或盐或衍生物的外消旋体)。替代性地,外消旋体(或外消旋前体)可与适当的光学活性化合物反应,例如乙醇,或在上述式中的任何一种的化合物含有酸性或碱性部分的情况下,与碱或酸如1-苯乙胺或酒石酸反应。所得的非对映体混合物可通过色谱法和/或分馏结晶法分离,并且通过技术人员熟知的方法将非对映体中的一者或两者转化为相应的纯对映体。上述式中的任何一种的手性化合物(及其手性前体)可以使用色谱法,通常是hplc,在不对称树脂上,用由含有0至50%体积的异丙醇(通常为2%至20%)以及0至5%体积的烷基胺(通常为0.1%的二乙胺)的碳氢化合物(通常是庚烷或己烷)组成的流动相,以对映体富集的形式获得。洗脱物的浓缩提供富集的混合物。可以采用亚临界和超临界流体的手性色谱法。可用于本公开的一些实施方案的手性色谱法的方法在本领域是已知的(见,例如,smith,roger m.,loughborough university,loughborough,uk;chromatographic science series(1998),75(supercritical fluid chromatography with packed columns),pp.223-249和其中引用的文献)。柱可以从chiral technologies,inc,west chester,pa.,usa,a subsidiary ofchemical industries,ltd.,tokyo,japan获得。
55.必需强调,上述式中的任何一种的化合物已以单一互变异构形式绘出,所有可能的互变异构形式包括在本公开的范围内。
56.如本文所用,术语“盐”或“盐类”指本发明的化合物的酸加成盐或碱加成盐。“盐类”特别包括“药学上可接受的盐”。术语“药学上可接受的盐”指保留本发明的化合物的生物有效性及性质的盐类,其通常不为生物学上或其他方面所不期望的,许多情况下,本发明的化合物能够利用氨基和/或羧基或与之类似的基团的存在,形成酸和/或碱盐类。
co.,easton,pennsylvania中找到。
66.如本文所用,术语“抑制”是指给定状况、症状、或病症或疾病的降低或抑制,或生物活性或过程的基线活性的显著下降。
67.术语“治疗”指逆转、减缓或抑制本文所描述疾病的进展。一些实施方案中,可在已发展出或观察到疾病的一种或多种体征或症状之后施用治疗(即治疗性处理)。其他实施方案中,可在没有疾病的体征或症状下施用治疗。例如,可在易感性受试者的症状发作之前施用治疗(即防治性处理)(例如,根据症状的病史和/或根据所暴露的病原菌)。亦可在症状解除之后继续治疗,例如,延缓或预防复发。
68.术语“状况”、“疾病”及“病症”可互换使用。
69.通常,本文所教导化合物的有效量因不同因素而异,如给定药物或化合物、药物配制剂、施用途径、疾病或病症类型、所治疗受试者或宿主的身份及类似因素,但可以由本领域技术人员常规地确定。本教导的化合物的有效量可以由本领域普通技术人员通过本领域常规方法容易地确定。
70.术语“有效量”意指当施用至受试者时造成有益或期望结果的量,该结果包括临床结果,例如,使所治疗受试者状况的症状相较于对照受到抑制、压制或降低。例如,有效量可以以单位剂量形式给予(例如,每天1mg至约50g,例如,每天1mg至约5g)。
71.术语本发明的化合物的“治疗有效量”指将引发受试者的生物或医学应答,例如,降低或抑制酶或蛋白质活性,或缓解症状、减轻状况、减慢或延缓疾病进展或预防疾病等等的本发明的化合物的量。一个非限制实施方案中,术语“治疗有效量”指当施用至受试者时,有效(1)至少部分减轻、抑制、防止和/或缓解(i)由hpk1介导、或(ii)与hpk1活性相关联、或(iii)表征为hpk1活性(正常或异常)的状况、病症或疾病;或(2)降低或抑制hpk1活性;或(3)降低或抑制hpk1表达;或(4)修改hpk1的蛋白质水平的本发明的化合物的量。另一个非限制实施方案中,术语“治疗有效量”指施用至细胞、或组织、或非细胞生物材料或介质时,有效至少部分降低或抑制hpk1活性;或部分或完全降低或抑制hpk1表达的本发明的化合物的量。
72.本文所描述的所有方法可以以任何合适顺序进行,除非本文中另有指示或其他清楚说明。使用本文所提供的任何实例及其他实例,或示例性语言(例如,“如”)仅意在更佳阐述本发明,除非另外声明,否则无意限制本发明范围。
73.上述式中所采用一般化学术语具有其一般定义。
74.如本文所用,术语“药学上可接受的载剂”包括任何及所有溶剂、分散介质、包衣、表面活性剂、抗氧化剂、防腐剂(例如,抗细菌剂、抗真菌剂)、等渗剂、延缓吸收剂、盐类、防腐剂、药物安定剂、结合剂、赋形剂、崩解剂、润滑剂、甜味剂、调味剂、染料及类似物及其组合,其如本领域技术人员所知(参见例如,remington’s pharmaceutical sciences,第18版,mack printing company,1990,pp.1289-1329)。除非任何常规载剂与活性成分不相容,否则考虑其用在治疗或药物组合物中。
75.此外,本发明的化合物,包括其盐,亦可他以其水合物的形式获得,或包括用于其结晶的其他溶剂。本发明的化合物可以固有地为溶剂合物或设计为与药学上可接受的溶剂(包括水)形成溶剂合物;因此本发明意在包括溶剂合物与非溶剂合物形式。术语“溶剂合物”指本发明的化合物(包括其药学上可接受的盐)与一个或多个溶剂分子形成的复合物。
此类溶剂分子是药物领域中常用的那些,其已知对接受者无害,例如,水、乙醇及类似物。术语“水合物”指其中溶剂分子为水的复合物。
76.本发明的化合物,包括其盐、水合物及溶剂合物,可以固有地或设计形成多晶型物。另一方面,本发明提供了包含本发明的化合物及药学上可接受的载剂的药物组合物。药物组合物可配制用于特定施用途径,如口服施用、肠胃外施用和直肠施用等。此外,本发明药物组合物可制成固体形式(包括但不限于胶囊、片剂、丸剂、粒剂、粉剂或栓剂),或呈液体形式(包括但不限于溶液、悬浮液或乳液)。药物组合物可以经受常规制药操作,如杀菌和/或可以含有常规的惰性稀释剂、润滑剂或缓冲剂,及佐剂,如防腐剂、安定剂、湿化剂、乳化剂及缓冲剂等。
77.典型地,药物组合物为片剂或明胶胶囊,其包含活性成分,连同a)稀释剂,例如,乳糖、右旋糖、蔗糖、甘露糖醇、山梨糖醇、纤维素和/或甘氨酸;b)润滑剂,例如,硅石、滑石、硬脂酸、其镁盐或钙盐和/或聚乙二醇;用于片剂还有c)结合剂,例如,硅酸镁铝、淀粉糊、明胶、黄芪胶、甲基纤维素、羧甲基纤维素钠和/或聚乙烯吡咯烷酮;若期望时,d)崩解剂,例如,淀粉、琼脂、藻酸或其钠盐,或泡腾混合物;和/或e)吸收剂、着色剂、调味剂和甜味剂。片剂可以根据本领域已知方法包被膜衣或肠衣。
78.用于口服施用的合适组合物包括有效量的本发明的化合物,其呈片剂、锭剂(lozenge)、水性或油性悬浮液、分散性粉剂或颗粒剂、乳液、硬或软胶囊、或糖浆或酏剂的形式。意在口服使用的组合物根据本领域已知的用于制造药物组合物的任何方法制备,并且此类组合物可以含有选自由以下组成的组中的一种或多种药剂:甜味剂、调味剂、着色剂及防腐剂,以提供药学上美观及适口的制剂。片剂可以含有与适合制造片剂的无毒性药学上可接受的赋形剂混合的活性成分。此类赋形剂为例如,惰性稀释剂如碳酸钙、碳酸钠、乳糖、磷酸钙或磷酸钠;造粒剂及崩解剂,例如,玉米淀粉或藻酸;结合剂,例如,淀粉、明胶或阿拉伯树胶(acacia);及润滑剂,例如硬脂酸镁、硬脂酸或滑石。片剂可以未包被或采用已知技术包被,以延缓在胃肠道中崩解及吸收,从而在较长时期内提供持续作用。例如,可采用延时材料,如单硬脂酸甘油酯或二硬脂酸甘油酯。用于口服使用的配制剂可以以硬明胶胶囊呈现,其中活性成分与惰性固体稀释剂例如,碳酸钙、磷酸钙或高岭土混合,或以软明胶胶囊呈现,其中活性成分与水或油介质,例如,花生油、液态石蜡或橄榄油混合。
79.某些可注射组合物为水性等渗溶液或悬浮液,并且栓剂有利地由脂肪乳液或悬浮液制备。所述组合物可以经过杀菌和/或包含佐剂,如防腐剂、安定剂、湿化剂或乳化剂、促进溶解剂、调节渗透压的盐和/或缓冲剂。此外,其亦可含有其他具有治疗价值的物质。所述组合物分别根据常规混合、造粒或包衣方法制备,并且含有约0.1-75%或含有约1-50%的活性成分。用于经皮应用的合适组合物包含有效量的本发明的化合物与合适的载剂。适用于经皮递送的载剂包括可吸收的药学上可接受的溶剂,以促进通过宿主皮肤。例如,经皮装置呈绷带形式,其包含背衬元件、含有化合物及任选地具有载剂的贮液器、任选地速率控制障壁,以便以控制且预定的速率长期递送化合物给宿主皮肤,并且打算固定该装置在皮肤上。
80.用于表面应用,例如至皮肤及眼睛的合适组合物,包括水性溶液、悬浮液、软膏剂、乳霜、凝胶或可喷洒配制剂,例如,采用气溶胶或类似物递送。此类表面递送系统将特别适合皮肤应用,例如用于皮肤癌症的治疗,例如,用于在防晒乳、洗液、喷雾及类似物中的防治
性用途。因此其特别适用于表面,包括本领域公知的配制剂,化妆品。其可以含有溶解剂、安定剂、张力增强剂、缓冲剂及防腐剂。
81.如本文所用,表面应用还可以适合吸入或鼻内应用。其可以方便地以干粉(单独、或作为混合物,例如与乳糖的干混物,或混合组分颗粒,与例如与磷脂形成)的形式从干粉吸入器递送,或使用或不使用合适推进剂下,呈气溶胶喷雾从加压容器、泵、喷瓶、雾化器或喷雾器递送。
82.本发明进一步提供包含本发明的化合物作为活性成分的无水药物组合物及剂型,因为水可能促进某些化合物的降解。
83.本发明的无水药物组合物及剂型可以使用无水或低水分的成分和在低水分及低湿度条件下制备。可以制备及储存无水药物组合物,因此维持其无水性质。因此,使用已知可以阻止暴露于水的材料包装无水组合物,以便其可包含在合适的配制盒内。合适包装的实例包括但不限于密封箔、塑料、单位剂量容器(例如,小瓶)、罩板包装及长条包装。
84.本发明进一步提供了药物组合物及剂型,其包含可以降低作为活性成分的本发明的化合物的降解速率的一种或多种药剂。此类药剂在本文中称为“安定剂”,包括但不限于,抗氧化剂如抗坏血酸、ph缓冲剂、或盐缓冲剂等。
85.3.化合物
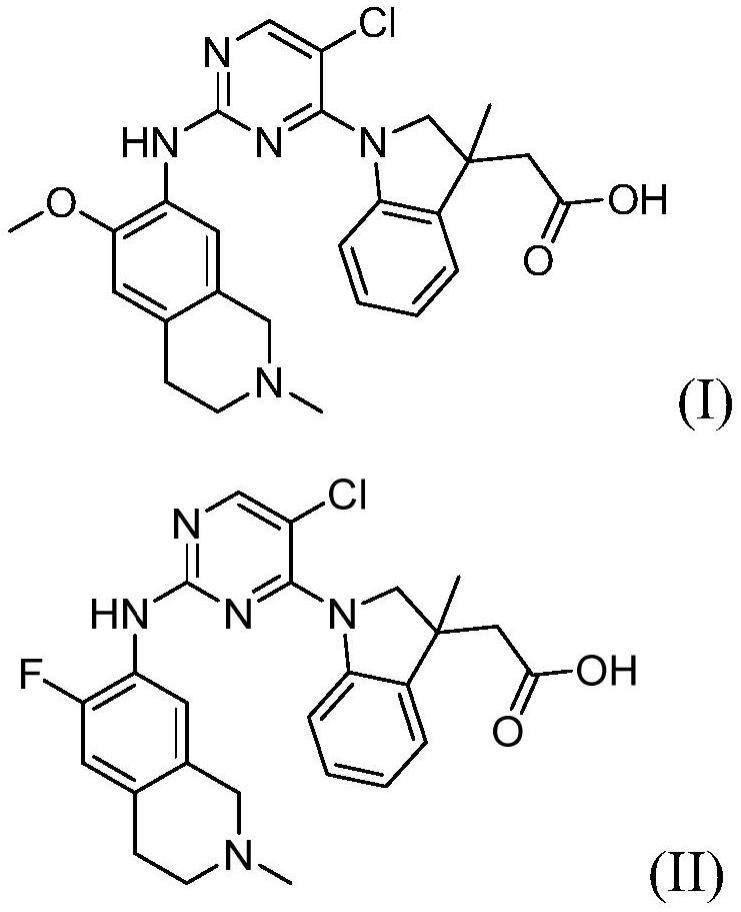
86.本公开的第一实施方案中,提供了由式i代表的化合物,
[0087][0088]
或其药学上可接受的盐,或立体异构体。
[0089]
本公开的第二实施方案中,提供了由式ia代表的化合物,
[0090][0091]
或药学上可接受的盐。
[0092]
本公开的第三实施方案中,提供了由式ib代表的化合物,
[0093]
[0094]
或药学上可接受的盐。
[0095]
本公开的第四实施方案中,提供了由式ii代表的化合物,
[0096][0097]
或其药学上可接受的盐,或立体异构体。
[0098]
本公开的第五实施方案中,提供了由式iia代表的化合物,
[0099]
或药学上可接受的盐。
[0100]
本公开的第六实施方案中,提供了由式iib代表的化合物,
[0101][0102]
或药学上可接受的盐。
[0103]
4.可治疗的疾病
[0104]
hpk1抑制剂、其药学上可接受的盐、其药物组合物可以用于调控(即抑制)hpk1活性的方法中,所述方法包括向有此需要的患者/受试者施用本文所描述的本发明的hpk1抑制剂化合物,或其药学上可接受的盐。
[0105]
特别地,本发明提供本发明的化合物,或其立体异构体、互变异构体、n-氧化物、水合物、溶剂合物和盐,特别是其药学上可接受的盐,或其混合物的用途,其用于治疗或防治疾病,特别是癌症(特别是造血性及实体肿瘤)或具有免疫应答失调的状况或其他与异常map4k1信号传导相关的病症。根据本发明的化合物的药学活性至少部分可以由其作为map4k1抑制剂的活性来解释。
[0106]
某些实施方案中,本发明的化合物或其药学上可接受的盐用于治疗性施用至有此需要的受试者,来治疗疾病或适应症,包括但不限于良性增生、动脉粥样硬化病症、败血症、自体免疫病症、心血管病症、病毒感染、神经变性病症、炎性病症及男性生育控制病症。
[0107]
某些实施方案中,本发明的化合物或其药学上可接受的盐用于治疗性施用,以在治疗癌症时,增强、刺激和/或增加免疫力。
[0108]
本发明的hpk1抑制剂化合物可以单独使用或与其他药剂或疗法联合使用,或作为
治疗疾病或病症(包括癌症)的佐剂或新辅助剂使用。
[0109]
某些实施方案中,本发明的方法可用于治疗癌症,包括但不限于骨癌、胰腺癌、皮肤癌、头颈部癌症、皮肤或眼内恶性黑色素瘤、子宫癌、卵巢癌、直肠癌、肛区癌症、胃癌、睾丸癌、子宫癌、输卵管癌、子宫内膜癌、子宫内膜癌、子宫颈癌、阴道癌、外阴癌、霍奇金病、非霍奇金淋巴瘤、食道癌、小肠癌、内分泌系统癌、甲状腺癌、甲状旁腺癌、肾上腺癌、软组织肉瘤、尿道癌、阴茎癌、慢性或急性白血病(包括急性髓性白血病、慢性髓性白血病、急性淋巴母细胞白血病、慢性淋巴细胞白血病)、儿童实体肿瘤、淋巴细胞性淋巴瘤、膀胱癌、肾脏或尿道癌、肾盂癌、中枢神经系统(cns)赘生物、原发性cns淋巴瘤、肿瘤新血管形成、脊髓轴肿瘤(spinal axis tumor)、脑干胶质瘤、垂体腺瘤、卡波西肉瘤、表皮样癌、鳞状细胞癌、t-细胞淋巴瘤、环境诱发的癌症(包括石棉诱发的那些),及上述癌症的组合。
[0110]
在一些实施方案中,可使用本发明的化合物治疗的癌症包括黑色素瘤(例如,转移性恶性黑色素瘤)、肾癌(例如,透明细胞癌)、前列腺癌(例如,激素难治性前列腺腺癌)、乳腺癌、三阴性乳腺癌、结肠癌及肺癌(例如,非小细胞肺癌及小细胞肺癌)。此外,可以使用本发明的化合物抑制其生长的难治性或复发性恶性肿瘤亦可治疗。
[0111]
在一些实施方案中,可使用本发明的化合物治疗的癌症包括但不限于实体肿瘤(例如,前列腺癌、结肠癌、食道癌、子宫内膜癌、卵巢癌、子宫癌、肾癌、肝癌、胰腺癌、胃癌、乳腺癌、肺癌、呼吸道、脑部癌症、眼部癌症、甲状腺及甲状旁腺癌症、皮肤癌、头颈部癌症、生殖器官癌症、消化道癌症、泌尿道癌症、胶质母细胞瘤、肉瘤、膀胱癌等)、血液癌症(例如,淋巴瘤,白血病如急性淋巴母细胞白血病(all)、急性骨髓性白血病(aml)、慢性淋巴细胞性白血病(cll)、慢性骨髓性白血病(cml)、dlbcl、套细胞淋巴瘤,非霍奇金淋巴瘤(包括复发或难治性nhl及复发性滤泡),霍奇金淋巴瘤或多发性骨髓瘤)、肉瘤,及其远距转移。
[0112]
在一些实施方案中,可使用本发明的化合物治疗的疾病及适应症包括但不限于血液癌症、肉瘤、肺癌、胃肠道癌症、泌尿生殖道癌症、肝脏癌症、骨癌、神经系统癌症、妇科癌症及皮肤癌。
[0113]
示例性血液癌症包括淋巴瘤及白血病,如急性淋巴母细胞白血病(all)、急性骨髓性白血病(aml)、急性早幼粒细胞性细胞白血病(apl)、慢性淋巴细胞性白血病(cll)、慢性骨髓性白血病(cml)、弥漫性大b-细胞淋巴瘤(dlbcl)、套细胞淋巴瘤、非霍奇金淋巴瘤(包括复发或难治性nhl及复发性滤泡)、霍奇金淋巴瘤、骨髓增生性疾病(例如,原发性骨髓纤维化(pmf)、真性红细胞增多症(pv)、原发性血小板增多症(et)、骨髓增生异常综合征(mds)、t-细胞急性淋巴母细胞淋巴瘤(t-all)、多发性骨髓瘤、皮肤t-细胞淋巴瘤、华氏巨球蛋白血症(waldenstrom’s macroglubulinemia)、毛细胞淋巴瘤、慢性髓性淋巴瘤及伯基特淋巴瘤。
[0114]
示例性肉瘤包括软骨肉瘤、尤文氏肉瘤(ewing’s sarcoma)、骨肉瘤、横纹肌肉瘤、血管肉瘤、纤维肉瘤、脂肪肉瘤、粘液瘤、横纹肌瘤、横纹肌肉瘤、纤维瘤、脂肪瘤、错构瘤和畸胎瘤。
[0115]
示例性肺癌包括非小细胞肺癌(nsclc)、小细胞肺癌、支气管癌(鳞状细胞、未分化小细胞、未分化大细胞、腺癌)、肺泡(细支气管)癌、支气管腺瘤、软骨错构瘤及间皮瘤。
[0116]
示例性胃肠癌包括食道(鳞状细胞癌、腺癌、平滑肌肉瘤、淋巴瘤)、胃(癌、淋巴瘤、平滑肌肉瘤)、胰腺(导管腺癌、胰岛瘤、胰高血糖素瘤、胃泌素瘤、类癌性肿瘤、血管活性肠
肽瘤(vipoma))、小肠(腺癌、淋巴瘤、类癌性肿瘤、卡波西肉瘤、平滑肌瘤、血管瘤、脂肪瘤、神经纤维瘤、纤维瘤)、大肠(腺癌、管状腺瘤、绒毛腺瘤、错构瘤、平滑肌瘤)的癌症及结直肠癌。
[0117]
示例性生殖泌尿道癌症包括肾脏(腺癌、威尔姆氏肿瘤(wilms’tumor)[肾母细胞瘤])、膀胱及尿道(鳞状细胞癌、移行细胞癌、腺癌)、前列腺(腺癌、肉瘤)和睾丸(精原细胞瘤、畸胎瘤、胚胎性癌、畸胎癌、绒毛膜癌、肉瘤、间质细胞癌、纤维瘤、纤维性瘤、类腺瘤、脂肪瘤)的癌症。
[0118]
示例性肝癌包括肝癌(肝细胞癌)、胆管癌、肝母细胞瘤、血管肉瘤、肝细胞腺瘤及血管瘤。
[0119]
示例性骨癌包括例如,骨原性肉瘤(骨肉瘤)、纤维肉瘤、恶性纤维性组织细胞瘤、软骨肉瘤、尤文氏肉瘤(ewing’s sarcoma)、恶性淋巴瘤(网状细胞肉瘤)、多发性骨髓瘤、恶性巨细胞肿瘤脊索瘤、骨软骨瘤(osteochronfroma)(骨软骨性外生骨疣)、良性软骨瘤、软骨母细胞瘤、软骨粘液性纤维肌瘤、骨样骨瘤及骨巨细胞瘤(giant cell tumor)。
[0120]
示例性神经系统癌症包括头骨(骨瘤、血管瘤、肉芽肿、黄瘤、变形性骨炎)、脑膜(脑膜瘤、脑膜肉瘤、神经胶质瘤)、脑(星细胞瘤、髓母细胞瘤(meduoblastoma)、神经胶质瘤、室管膜细胞瘤、胚细胞瘤(松果体瘤)、胶质母细胞瘤、多形性胶质母细胞瘤、少突神经胶质瘤、许旺细胞瘤(schwannoma)、视网膜母细胞瘤、天生性肿瘤)和脊髓(神经纤维瘤、脑膜瘤、神经胶质瘤、肉瘤)的癌症,以及神经母细胞瘤及发育不良性节细胞瘤(lhermitte-duclos disease)。
[0121]
示例性妇科癌症包括子宫(子宫内膜癌)、子宫颈(子宫颈癌、肿瘤前的子宫颈发育不良)、卵巢(卵巢癌(浆液性囊状腺癌、粘液性囊状腺癌、未分类癌)、粒层细胞-鞘细胞肿瘤、支持间质细胞(sertoli-leydig cell)肿瘤、无性细胞瘤、恶性畸胎瘤)、外阴(鳞状细胞癌、上皮内癌、腺癌、纤维肉瘤、黑色素瘤)、阴道(透明细胞癌、鳞状细胞癌、葡萄状肉瘤(胚胎性横纹肌肉瘤)及输卵管(癌,carcinoma)的癌症。
[0122]
示例性皮肤癌症包括黑色素瘤、基底细胞癌、鳞状细胞癌、卡波西肉瘤、梅克尔细胞皮肤癌、再生不良性痣、脂肪瘤、血管瘤、皮肤纤维肌瘤及蟹足肿。在一些实施方案中,可使用本发明的化合物治疗的疾病及适应症包括(但不限于)镰状细胞疾病(例如,镰状细胞贫血)、三阴性乳腺癌(tnbc)、骨髓增生不良综合征、睾丸癌、胆道癌、食道癌和泌尿道上皮癌。
[0123]
示例性头颈部癌症包括胶质母细胞瘤、黑色素瘤、横纹肌肉瘤、淋巴肉瘤、骨肉瘤、鳞状细胞癌、腺癌、口癌、喉癌、鼻咽癌、鼻与鼻旁癌症,及甲状腺与甲状旁腺癌症。
[0124]
在一些实施方案中,该hpk1抑制剂可用于治疗产生pge2的肿瘤(例如,过表达cox-2的肿瘤)和/或腺苷的肿瘤(过表达cd73及cd39的肿瘤)。已在许多种肿瘤中检测到过表达的cox-2,如结直肠、乳房、胰脏及肺脏等癌症,其与预后不佳相关。已在血液癌症模式中(如raji(伯基特淋巴瘤,burkitt’s lymphoma)及u937(急性前单核细胞白血病))以及患者的胚细胞中报告过cox-2的过表达。cd73在各种不同人癌中上调,包括那些结肠、肺、胰脏及卵巢的癌症。cd73的较高表达水平已与肿瘤新血管形成、侵袭性及转移相关,并与较短的乳腺癌患者存活期有关。
[0125]
可治疗乳腺癌的实例包括但不限于三阴性乳腺癌、浸润性导管癌、浸润性小叶癌、
原位导管癌及原位小叶癌。
[0126]
呼吸道癌症的实例包括但不限于小细胞及非小细胞肺癌,以及支气管腺瘤及胸膜肺母细胞瘤。
[0127]
可治疗脑部癌症的实例包括但不限于脑干及下丘脑胶质瘤、小脑及大脑星形细胞瘤、胶质母细胞瘤、髓母细胞瘤、室管膜瘤,以及神经外胚层及松果体肿瘤。
[0128]
可治疗的男性生殖器官肿瘤包括但不限于前列腺及睾丸的癌症。
[0129]
可治疗的女性生殖器官肿瘤包括但不限于子宫内膜癌、子宫颈癌、卵巢癌、阴道癌及外阴癌,以及子宫肉瘤。
[0130]
可治疗的卵巢癌症包括但不限于浆液性肿瘤、子宫内膜样肿瘤、粘液性囊腺癌、颗粒细胞肿瘤、支持-间质细胞(sertoli-leydig cell)肿瘤及男性细胞瘤(arrhenoblastoma)。
[0131]
可治疗的子宫颈癌症包括但不限于鳞状细胞癌、腺癌、腺鳞状癌、小细胞癌、神经内分泌肿瘤、玻璃状细胞癌及子宫颈乳头状腺癌。
[0132]
可治疗的消化道肿瘤包括但不限于肛门、结肠、结直肠、食道、膀胱、胃、胰脏、直肠、小肠及唾液腺的癌症。
[0133]
可治疗的食道癌症包括但不限于食道细胞癌及腺癌,以及鳞状细胞癌、平滑肌肉瘤、恶性黑色素瘤、横纹肌肉瘤及淋巴瘤。
[0134]
可治疗的胃部癌症包括但不限于肠道型及扩散型胃腺癌。
[0135]
可治疗的胰脏癌症包括但不限于胰管腺癌、腺鳞状癌及胰脏内分泌肿瘤。
[0136]
可治疗的尿路肿瘤包括但不限于膀胱、阴茎、肾脏、肾盂、输尿管、尿道及人乳突肾癌症。
[0137]
可治疗的肾脏癌症包括但不限于肾细胞癌、泌尿道上皮细胞癌、近肾小球细胞肿瘤(肾素瘤)、血管平滑肌脂肪瘤、肾腮腺嗜酸性颗粒细胞瘤、毕氏管(bellini duct)癌、肾脏的透明细胞肉瘤、中胚叶肾瘤及威尔姆氏肿瘤(wilms’tumor)。
[0138]
可治疗的膀胱癌症包括但不限于移行细胞癌、鳞状细胞癌、腺癌、肉瘤及小细胞癌。
[0139]
可治疗的眼部癌症包括但不限于眼内黑色素瘤及视网膜母细胞瘤。
[0140]
可治疗的肝脏癌症包括但不限于肝细胞癌(出现或不出现纤维薄层型变异的肝脏细胞癌)、胆管癌(肝内胆管癌)及混合肝细胞胆管癌。
[0141]
可治疗的皮肤癌症包括但不限于鳞状细胞癌、卡波西肉瘤、恶性黑色素瘤、梅克尔细胞皮肤癌及非黑色素瘤皮肤癌。
[0142]
可治疗的头颈部癌症包括(但不限于)头颈部鳞状细胞癌、喉癌、下咽癌、鼻咽癌、口咽癌、唾液腺癌、唇及口腔癌症及鳞状细胞。
[0143]
可治疗的淋巴瘤包括但不限于aids-相关淋巴瘤、非霍奇金淋巴瘤、皮肤t-细胞淋巴瘤、伯基特淋巴瘤、霍奇金疾病及中枢神经系统淋巴瘤。
[0144]
可治疗的肉瘤包括但不限于软组织的肉瘤、骨肉瘤、恶性纤维性组织细胞瘤、淋巴肉瘤及横纹肌肉瘤。
[0145]
可治疗的白血病包括但不限于急性髓性白血病、急性淋巴母细胞性白血病、慢性淋巴细胞性白血病、慢性骨髓性白血病及毛细胞白血病。
[0146]
某些实施方案中,本发明的化合物可以用于治疗涉及map4k1的各种不同其他疾病,如心血管与肺部疾病。
[0147]
某些实施方案中,本发明的化合物可以用于供治疗和/或防治心血管、炎性及纤维化病症、肾病症,特别是急性及慢性肾功能不全,及急性及慢性肾衰竭的药物。
[0148]
本文中,术语“肾功能不全”包括肾功能不全的急性及慢性两种临床表现,及潜在或相关的肾病症,如糖尿病及非糖尿病性肾病、高血压性肾病、缺血性肾病症、肾灌注不足、透析中低血压、阻塞性尿路病、肾狭窄、肾小球病、肾小球肾炎(如,例如,原发性肾小球肾炎;微小病变型肾小球肾炎(类脂性肾病);膜性肾小球肾炎;局灶节段性肾小球硬化症(fsgs);膜-增生性肾小球肾炎;急进性肾小球肾炎;环间膜增生性肾小球肾炎(iga肾炎、柏格氏病(berger’s disease));感染后肾小球肾炎;续发性肾小球肾炎)、糖尿病、系统性红斑狼疮、类淀粉沉积症、古德帕斯丘综合征(goodpasture syndrome)、韦氏肉芽肿病(wegener’s granulomatosis)、过敏性紫斑症(henoch-schonlein purpura)、显微性多血管炎、急性肾小球肾炎、肾盂肾炎(例如,因尿石症、良性前列腺肥大、糖尿病、畸形、滥用止痛药、克隆氏症(crohn's disease)所致者)、肾小球硬化症、肾脏的小动脉坏死(arteriolonecrose)、肾小管间质性疾病、肾病病症如原发性及先天性或后天性肾病症、奥尔波特综合征(alport syndrome)、肾炎、免疫性肾脏病症如肾脏移植排斥及免疫复合体诱的肾病症、毒性物质诱发的肾病、显影剂诱发的肾病、糖尿病及非糖尿病性肾病、肾囊肿、肾硬化、高血压性肾硬化及肾病综合征,其可以经诊断表征例如,通过肌酸酐和/或排水量异常减少、血中尿素、氮、钾和/或肌酸酐等浓度异常升高、肾酶活性改变(例如,谷酰基合成酶)、尿液渗透压或尿液体积改变、微白蛋白尿升高、巨量白蛋白尿、肾小球及小动脉损伤、肾管扩张、高磷酸盐血症和/或需要洗肾。
[0149]
某些实施方案中,本发明的化合物可以用于治疗和/或防治肾功能不全的后遗症,例如,肺水肿、心脏衰竭、尿毒症、贫血、电解质紊乱(例如,高血钙症、低血钠症)及骨与碳水化合物代谢紊乱。
[0150]
某些实施方案中,本发明的化合物可以用于治疗和/或防治肾功能不全的后遗症,例如,肺水肿、心脏衰竭、尿毒症、贫血、电解质紊乱(例如,高血钙症、低血钠症)及骨与碳水化合物代谢紊乱。
[0151]
某些实施方案中,本发明的化合物进一步适用于治疗和/或预防多囊性肾脏疾病(pckd)及adh分泌不当综合征(siadh)。
[0152]
某些实施方案中,本发明的化合物亦适用于治疗和/或防治代谢综合征、高血压、顽固性高血压、急性和慢性心脏衰竭、冠心病、稳定性和不稳定性心绞痛、外周和心脏血管病症、心律不整、心房和心室心律不整以及传导功能受损,例如,第l-lll级房室传导阻滞(ab阻滞l-lll)、上心室心搏过速、心房纤维性颤动、心房扑动、心室纤维性颤动、心室扑动、心室心搏过速、尖端扭转型心搏过速、心房及心室额外收缩、av-交界区性额外收缩、病窦综合征、昏厥、av-结回旋心搏过速、wolff-parkinson-white综合征、急性冠脉综合征(acs)、自体免疫心脏病症(心包膜炎、心内膜炎、心瓣膜炎、主动脉炎、心肌病)、休克(如心源性休克、败血性休克及过敏性休克)、动脉瘤、拳师犬心肌病(boxer-cardiomyopathy)(心室提前收缩(pvc));用于治疗和/或防治血栓栓塞性病症及缺血症如心肌缺血、心肌梗塞、中风、心脏肥大、暂时性和缺血性发作、子痫前症、炎性心血管病症、冠状动脉及外周动脉痉挛、水肿
形成(例如,肺水肿、脑水肿、肾水肿或因心脏衰竭、外周循环紊乱、再灌注损伤、动脉与静脉血栓造成的水肿)、心肌功能不全、血管内皮功能障碍;以预防再狭窄(例如,在血栓溶解疗法、经皮经腔血管成形术(pta)、经腔冠状动脉血管成形术(ptca)、心脏移植和旁路手术之后),以及微血管与大血管损伤(脉管炎)、增加的纤维蛋白原及低密度脂蛋白(ldl)水平及增加的血纤蛋白溶解酶原激活物抑制剂1(pai-1)浓度,并且还用于治疗和/或防治勃起功能障碍和女性性功能障碍。
[0153]
某些实施方案中,本发明的化合物亦适用于治疗和/或防治气喘病症、肺动脉高血压(pah)及肺高血压(ph)的其他形式,包括左心病、hiv、镰状细胞贫血、血栓栓塞(cteph)、结节病、copd或肺纤维化相关的肺高血压、慢性阻塞性肺病(copd)、急性呼吸窘迫综合征(ards)、急性肺损伤(ali)、α-1-抗胰蛋白酶缺陷(aatd)、肺纤维化、肺气肿(例如,因香烟烟雾诱发的肺气肿)及囊性纤维化(cf)。
[0154]
某些实施方案中,本发明的化合物亦可有效用于控制表征为no/cgmp系统紊乱的中枢神经系统病症。其特别适用于在认知损害,如特别与以下情况/疾病/综合征相关联发生的那些之后改善知觉、注意力、学习或记忆:诸如轻度认知损害、年龄相关性学习及记忆损害、年龄相关性记忆丧失、血管性痴呆、脑外伤、中风、中风后发生的痴呆(中风后痴呆)、创伤后脑外伤、一般专注力损害、儿童专注力损害伴学习及记忆问题、阿尔茨海默病、路易体(lewy body)痴呆、额叶变性痴呆,包括皮克综合征、帕金森病、皮质基底节变性进行性痴呆、肌萎缩性侧索硬化症(als)、亨廷顿病、脱髓鞘、多发性硬化、丘脑变性、克雅氏痴呆(creutzfeld-jacob dementia)、hiv痴呆、精神分裂症伴痴呆或科尔萨科夫精神病(korsakoff’s psychosis)。
[0155]
某些实施方案中,本发明的化合物亦适用于治疗和/或防治中枢神经系统病症,如焦虑、紧张及抑郁的状态、cns-相关的性功能障碍及睡眠紊乱,及用于控制在摄取食物、兴奋剂和成瘾物的病理性紊乱。
[0156]
某些实施方案中,本发明的化合物进一步适用于控制脑部血流,因此代表控制偏头痛的有效药剂。
[0157]
某些实施方案中,本发明的化合物亦适用于防治和控制脑梗塞(脑中风)的后遗症,如中风、脑缺血及脑外伤。根据本发明的化合物同样可用于控制疼痛及耳鸣状态。
[0158]
某些实施方案中,本发明的化合物具有抗炎作用,因此可作为抗炎剂,用于治疗和/或防治败血病(sirs)、多器官衰竭(mods、mof)、肾脏的炎性病症、慢性肠部炎症(ibd,克隆氏症(crohn's disease)、uc)、胰脏炎、腹膜炎、类风湿病症、炎性皮肤病症及炎性眼部病症。
[0159]
某些实施方案中,本发明的化合物亦可以用于治疗和/或防治自体免疫疾病。
[0160]
某些实施方案中,本发明的化合物亦适用于治疗和/或防治内脏器官的纤维化病症,例如,肺脏、心脏、肾脏、骨髓及特别是肝脏,及皮肤纤维化及纤维化眼部病症。
[0161]
如本文所用,术语“纤维化病症”特别地包括下列:肝纤维化、肝硬化、肺纤维化、心肌纤维化、肾病、肾小球肾炎、间质性肾纤维化、因糖尿病造成的纤维化损害、骨髓纤维化及类似的纤维化病症、硬皮病、硬斑病、蟹足肿、增生性疤痕(亦继手术过程之后)、痣、糖尿病视网膜病、增生性玻璃体视网膜病及结缔组织病症(例如,结节病)。
[0162]
某些实施方案中,本发明的化合物亦适用于控制手术后瘢痕,例如,因青光眼手术
造成的瘢痕。
[0163]
某些实施方案中,本发明的化合物亦可以用于老化及角质化皮肤的美容。
[0164]
某些实施方案中,本发明的化合物适用于治疗和/或防治肝炎、赘生物、骨质疏松症、青光眼和胃轻瘫。
[0165]
某些实施方案中,本发明的化合物适用于治疗和/或防治病毒感染(例如,hiv及卡波西肉瘤);炎性及自体免疫疾病(例如,结肠炎、关节炎、阿尔茨海默病、肾小球肾炎和伤口愈合);细菌、真菌、和/或寄生物感染;皮肤疾病(例如,银屑病);基于过度增生的疾病,其表征为细胞(例如,纤维母细胞、肝细胞、骨和骨髓细胞、软骨或平滑肌细胞或上皮细胞)的数量增加(例如,子宫内膜增生);骨疾病及心血管疾病(例如,再狭窄及肥大)。
[0166]
在另一个实施方案中,本发明的化合物亦可以用于治疗或预防妇女的子宫纤维瘤(子宫平滑肌瘤或子宫肌瘤)。子宫纤维瘤为子宫肌膜(为子宫的平滑肌层)的良性肿瘤。子宫纤维瘤在女人生命中慢慢生长,其生长依赖于女性性激素雌二醇及孕酮。因此,约70%及》80%的白人及非裔美国妇女的子宫纤维瘤的好发高峰分别在35岁到更年期之间,之后因激素量减少即会萎缩。约30%及45%的白人及非裔美国妇女分别因其纤维瘤而出现临床相关症状,其是与月经周期相关的大量经血及疼痛(david等人,eur j obstet gynecol reprod biol.199:137-140,2016)。此时的大量经血是界定在月经周期中流血超过80ml。子宫纤维瘤的粘膜下位置(例如,直接位于子宫内膜下的那些)似乎对子宫流血的影响甚至更严重,可能造成受影响妇女贫血。此外,会因此随子宫纤维瘤的症状,严重影响受影响妇女的生活品质。
[0167]
某些实施方案中,本发明的化合物用于治疗和/或防治慢性肾病症、急性和慢性肾功能不全、糖尿病、炎性或高血压性肾病、纤维化病症、心功能不全、心绞痛、高血压、肺高血压、缺血、血管病症、血栓栓塞性病症、动脉硬化、镰状细胞贫血、勃起功能障碍、良性前列腺增生、与良性前列腺增生相关的排尿困难、亨廷顿病、痴呆、阿尔茨海默病和克雅氏病。
[0168]
本发明提供了用于治疗和/或防治慢性肾病症、急性和慢性肾功能不全、糖尿病、炎性或高血压性肾病、纤维化病症、心脏功能不全、心绞痛、高血压、肺高血压、缺血、血管病症、血栓栓塞性病症、动脉血管硬化、镰状细胞贫血、勃起功能障碍、良性前列腺肥大、与良性前列腺肥大相关的排尿困难、亨廷顿病、痴呆、阿尔茨海默病和克雅氏病的方法。
[0169]
本发明进一步提供了根据本发明的化合物用于治疗和/或防治病症,特别是上述病症的用途。
[0170]
本发明进一步提供了使用有效量的根据本发明的化合物中的至少一种来治疗和/或防治病症,特别是上述病症的方法。
[0171]
因此,本发明的化合物可以用于通过外源性和/或内源性配体来抑制、阻断、减少、或降低map4k1活化,而降低肿瘤生长及调控失调的免疫应答,例如,在癌症及癌症免疫疗法的背景下,用于阻断免疫抑制性,及提高免疫细胞活化与浸润。此方法包括向有此需要的哺乳动物(包括人)施用本发明的化合物量或其药学上可接受的盐、异构体、多形物、代谢物、水合物、溶剂合物或酯以治疗该病症。
[0172]
本发明亦提供了治疗涉及map4k1的各种不同其他病症的方法,诸如但不限于病症伴失调免疫应答、炎症、针对感染与癌症的疫苗接种、病毒感染、肥胖和膳食诱发的肥胖、脂肪增多、代谢病症、肝脂肪变性和子宫纤维瘤。这些病症已在人中良好表征,但亦在其他哺
乳动物中具有类似病因,并且可以通过施用本发明的药物组合物来治疗。
[0173]
5.联合疗法
[0174]
本发明的化合物可以用于与适用于治疗可利用该主题化合物治疗的疾病或适应症的一种或多种额外/第二治疗剂的联合疗法。
[0175]
因此某些实施方案中,例如,使用本发明化合物的本发明方法可以包括向有此需要的受试者施用进一步的治疗剂。该进一步的治疗剂可以为:(i)阻断或抑制免疫系统检查点的免疫调控剂,该检查点可能是或可能不是nf-κb途径的组分;和/或(ii)直接刺激免疫效应子反应的药剂,如细胞因子、或肿瘤特异性过继性转移t细胞群、或针对肿瘤细胞所表达蛋白质的特异性抗体;和/或(iii)包含肿瘤抗原或其免疫原性片段的组合物;和/或(iv)化疗剂。
[0176]
某些实施方案中,该第二治疗剂包括pi3k-akt-mtor途径的抑制剂、raf-mapk途径的抑制剂、jak-stat途径的抑制剂、β连环蛋白(catenin)途径的抑制剂、notch途径的抑制剂、刺猬信号途径的抑制剂、pim激酶的抑制剂、和/或伴侣蛋白蛋白质与细胞周期进展的抑制剂。某些实施方案中,本发明的联合疗法可降低细胞群出现抗药性的可能性,和/或降低治疗的毒性。
[0177]
某些实施方案中,本发明的hpk1抑制剂化合物可以与下列激酶的一种或多种抑制剂组合,用于治疗癌症:akt1、akt2、akt3、tgf-βρν、pka、pkg、pkc、cam-激酶、磷酸化酶激酶、mekk、erk、mapk、mtor、egfr、her2、her3、her4、ins-r、igf-1r、ir-r、pdgfαr、pdgfβr、csfir、kit、flk-ii、kdr/flk-1、flk-4、flt-1、fgfr1、fgfr2、fgfr3、fgfr4、c-met、ron、sea、trka、trkb、trkc、flt3、vegfr/flt2、flt4、ephal、epha2、epha3、ephb2、ephb4、tie2、src、fyn、lck、fgr、btk、fak、syk、frk、jak、abl、alk和b-raf。
[0178]
某些实施方案中,本发明hpk1抑制剂化合物可以与下列抑制剂中的一种或多种组合,用于治疗癌症,包括:fgfr抑制剂(fgfr1、fgfr2、fgfr3或fgfr4,例如,azd4547、bay 1187982、arq087、bgj398、bibf1120、tki258、德立替尼(lucitanib)、多韦替尼(dovitinib)、tas-120、jj-42756493、debiol347、incb54828、incb62079和incb63904)、jak抑制剂(jak1和/或jak2,例如,鲁索利替尼(ruxolitinib)、巴瑞替尼(baricitinib)、或伊他替尼(itacitinib)(incb39110))、ido抑制剂(例如,艾卡哚司他(epacadostat)和nlg919)、lsd1抑制剂(例如,gsk2979552、incb59872及incb60003)、tdo抑制剂、pi3k-δ抑制剂(例如,incb50797及incb50465)、pi3k-γ抑制剂(如pi3k-γ选择性抑制剂)、csf1r抑制剂(例如,plx3397及ly3022855)、tam受体酪氨酸激酶(tyro-3、axl和mer)、芳基烃受体(ahr)调控剂(如拉喹莫德(laquinimod)、氨基黄酮、cb7993113、ch223191、6,2’,4
’‑
三甲氧基黄酮(tmf)、gnf351(n-(2-(1h-吲哚-3-基)乙基)-9-异丙基-2-(5-甲基吡啶-3-基)-9h-嘌呤-6-胺)、氨基黄酮、nki150460、吲哚-3-甲醇、β-萘啶黄酮及其二聚体、二吲哚基甲烷(dim)、4-羟基泰莫西芬(tamoxifen)、来氟米特(leflunomide)、雷洛昔芬(raloxifene)、曲尼司特(tranilast)、氟他胺(flutamide)、美西律(mexiletine)、尼莫地平(nimodiphine)、奥美拉唑(omeprazole)、舒林酸(sulindac)、曲尼司特(tranilast)和tcdd(2,3,7,8-四氯二苯并-p-二恶英))、血管生成抑制剂、白介素受体抑制剂、含溴与额外末端家族成员抑制剂(例如,溴域抑制剂或bet抑制剂,如otx015、cpi-0610、incb54329和incb57643)和腺苷受体拮抗剂或其组合。
[0179]
某些实施方案中,本发明的hpk1抑制剂化合物可以与hdac的抑制剂,如帕比司他(panobinostat)及伏立诺他(vorinostat)组合。
[0180]
某些实施方案中,本发明的hpk1抑制剂化合物可以与c-met的抑制剂,如昂纳珠单抗(onartumzumab)、替万提尼(tivantnib)和卡马替尼(capmatinib)(inc-280)组合。
[0181]
某些实施方案中,本发明的hpk1抑制剂化合物可以与btk的抑制剂,如依鲁替尼(ibrutinib)组合。
[0182]
某些实施方案中,本发明的hpk1抑制剂化合物可以与mtor的抑制剂,例如,雷帕霉素(rapamycin)、西罗莫司(sirolimus)、特罗莫司(temsirolimus)和依维莫司(everolimus)组合。
[0183]
某些实施方案中,本发明的hpk1抑制剂化合物可以与mek的抑制剂,如曲美替尼(trametinib)、司美替尼(selumetinib)及gdc-0973组合。
[0184]
某些实施方案中,本发明的hpk1抑制剂化合物可以与hsp90的抑制剂(例如,坦螺旋霉素(tanespimycin))、周期蛋白依赖性激酶的抑制剂(例如,帕博西尼(palbociclib))、parp的抑制剂(例如,奥拉帕尼(olaparib))及pim激酶的抑制剂(lgh447、incb053914和sgi-1776)组合。
[0185]
某些实施方案中,本发明的hpk1抑制剂化合物可以与dna传感器(c-gas)和/或其下游转接蛋白sting的激动剂组合。
[0186]
cgas(环状gmp-amp合成酶)-sting(干扰素基因的刺激剂)途径为先天免疫系统的组分,其功能在于检测细胞溶质dna的存在,且应答触发炎性基因的表达,造成衰老或激活防御机制。dna从正常核位置移至细胞溶质与肿瘤发生或病毒感染相关。已在细胞溶质中发现cgas,且当直接结合细胞溶质dna时,cgas会形成二聚体,催化由atp及gtp产生2
’3’‑
cgamp。所得cgamp即具有第二信使的作用,结合sting并触发激活转录因子irf3。活化irf3造成第1型ifn-β转录,使许多下游靶基因开始进行多样生物反应,如病毒应答、肿瘤侦测、自体免疫及细胞衰老。许多肿瘤细胞中,构成性活性dna损伤反应造成细胞质dna累积及激活cgas/sting途径。已在淋巴瘤细胞中显示,nkg2d配体rae1会以依赖sting/irf3的方式上调,因此有助于nk介导的肿瘤清除。在抗原呈递细胞(如树突状细胞)中激活c-gas-sting途径已显示增强其功能及加强抗肿瘤免疫力。
[0187]
某些实施方案中,本发明的hpk1抑制剂化合物可以与一种或多种免疫检查点抑制剂组合。
[0188]
效应子t细胞活化作用通常被mhc复合物所呈递的tcr识别的抗原性肽触发。然后由刺激的信号与抑制效应子t细胞应答的信号之间的平衡来决定所达成的活化类型与程度。如本文所用,“免疫系统检查点”是指任何可改变平衡状态而倾向抑制效应子t细胞应答的分子相互作用。亦即当发生该分子相互作用时,负向调节效应子t细胞的活化。此类相互作用可能为直接性,如配体与细胞表面受体间的相互作用传递抑制信号给效应子t细胞。或其可能为间接性,如阻断或抑制配体与细胞表面受体间的相互作用传递活化信号给效应子t细胞,或该相互作用促进抑制性分子或细胞的止调,或被效应子t细胞所需要的代谢物的酶消减,或其任何组合。
[0189]
免疫系统检查点的实例包括:a)吲哚胺2,3-二氧合酶(ido1与其底物之间的相互作用;b)pd1与pd-l1和/或pd1与pd-l2之间的相互作用;c)ctla-4与cd86和/或ctla-4与
cd80之间的相互作用;d)b7-h3和/或b7-h4与其各自配体之间的相互作用;e)hvem与btla之间的相互作用;f)gal9与tim3之间的相互作用;g)mhc i或ii类与lag 3之间的相互作用;及h)mhc i或ii类与kir之间的相互作用;i)ox40(cd134)与ox40l(cd252)之间的相互作用;j)cd40与cd40l(cd154)之间的相互作用;k)4-1bb(cd137)与配体(包括4-1bbl)之间的相互作用;l)gitr与配体(包括gitrl)之间的相互作用。
[0190]
因此示例性免疫检查点抑制剂包括针对以下免疫检查点分子的抑制剂,如cd20、cd27、cd28、cd39、cd40、cd 122、cd96、cd73、cd47、ox40、gitr、csf1r、jak、pi3kδ、pi3kγ、tam、精氨酸酶、cd137(亦已知为4-1bb)、icos、a2ar、b7-h3、b7-h4、btla、ctla-4、lag3、tim3、vista、pd-1、pd-l1和pd-l2。
[0191]
针对本发明目的的代表性检查点为检查点(b),亦即pd1与其任一配体pd-l1及pd-l2间的相互作用。pd1表达在效应子t细胞上。与其任一配体接合所产生的信号下调活化作用。有些肿瘤表现该等配体。许多实体肿瘤(包括黑色素瘤)特别表达pd-l1。因此这些肿瘤通过t细胞的抑制性pd-1受体的活化来下调免疫介导的抗肿瘤效应。通过阻断pd1与其一种或两种配体之间的相互作用,可移除免疫应答的检查点,造成增大的抗肿瘤t细胞应答。因此,pd1及其配体即为免疫系统检查点组分的实例,可能成为本发明方法的目标。
[0192]
针对本发明目的的另一个检查点为检查点(c),亦即t细胞受体ctla-4与其配体(b7蛋白质(b7-1及b7-2))之间的相互作用。t细胞表面在开始活化后,通常上调ctla-4,且与配体结合时,抑制进一步/继续活化。ctla-4与亦表达在t细胞表面的受体cd28竞争结合至b7蛋白质,但上调活化作用。因此通过阻断与b7蛋白质的ctla-4相互作用,但不阻断cd28与b7蛋白质的相互作用,可移除免疫应答的其中一个正常检查点,造成增大的抗肿瘤t细胞应答。因此,ctla-4及其配体即为免疫系统检查点组分的实例,可能成为本发明方法的目标。
[0193]
在一些实施方案中,免疫检查点分子为选自cd27、cd28、cd40、icos、ox40、gitr及cd137的刺激性检查点分子。
[0194]
在一些实施方案中,免疫检查点分子为选自a2ar、b7-h3、b7-h4、btla、ctla-4、ido、kir、lag3、nox2、pd-1、tiμ3、siglec7、siglec9和vista的抑制性检查点分子,和其结合配偶(如pd-l1和pd-l2)。
[0195]
在一些实施方案中,本文所提供化合物可以联合选自以下的一种或多种药剂使用:kir抑制剂、tigit抑制剂、lair1抑制剂、cd 160抑制剂、2b4抑制剂及tgfrβ抑制剂。
[0196]
在一些实施方案中,本文所提供化合物可以联合免疫检查点抑制剂使用,其通常为小型有机分子的小分子抑制剂(smi)。例如,某些实施方案中,ido1的抑制剂包括艾卡哚司他(epacadostat)(incb24360)、吲哚莫德(indoximod)、gdc-0919(nlg919)及f001287。ido1的其他抑制剂包括1-甲基色氨酸(1mt)。
[0197]
在一些实施方案中,免疫检查点分子的抑制剂亦称为“免疫调控剂”,其包括施用受试者时阻断或抑制免疫系统检查点的作用的任何药剂,在受试者中造成上调免疫效应子反应,通常为t细胞效应子反应,其可能包括抗肿瘤t细胞效应子反应。
[0198]
本发明方法所使用的免疫调控剂可阻断或抑制上述任何免疫系统检查点。该药剂可能为抗体或可造成所述阻断或抑制的任何其他合适药剂。因此该药剂通常称为所述检查点的抑制剂。
[0199]
如本文所用,“抗体”包括完整抗体及其任何抗原结合片段(亦即“抗原结合部分”)或单链。抗体可为多克隆抗体或单克隆抗体,且可能采用任何合适方法制造。术语抗体的“抗原结合部分”所涵盖的结合片段实例包括fab片段、f(ab’)2片段、fab’片段、fd片段、fv片段、dab片段,及分离的互补决定区(cdr)。单链抗体(如scfv)及重链抗体(如vhh及骆驼抗体)亦旨在涵盖在术语抗体的“抗原结合部分”内。
[0200]
某些实施方案中,本发明hpk1抑制剂所使用的免疫调控剂为抗pd1抗体、抗pd-ll抗体、抗pd-l2抗体或抗ctla-4抗体。
[0201]
在一些实施方案中,免疫检查点分子的抑制剂为pd-1的抑制剂,例如,抗pd-1单克隆抗体。在一些实施方案中,抗pd-1单克隆抗体为纳武单抗(nivolumab)(mdx-1106)、帕博利珠单抗(pembrolizumab)(merck 3475或拉博利珠单抗(lambrolizumab))、匹地利珠单抗(pidilizumab)(ct-011)、替雷利珠单抗(tislelizumab)(bgb-a317)、卡瑞利珠单抗(camrelizumab)(shr-1210)、斯巴达珠单抗(spartalizumab)(pdr001)、或amp-514(medi0680)。在一些实施方案中,抗pd-1单克隆抗体为纳武单抗(nivolumab)或帕博利珠单抗(pembrolizumab)。在一些实施方案中,抗pd1抗体为帕博利珠单抗(pembrolizumab)。在一些实施方案中,抗pd-1抗体为卡瑞利珠单抗(camrelizumab)(shr-1210)。某些实施方案中,pd-1的抑制剂为amp-224(会与pd-1结合的pd-l2fc融合蛋白质)或aunp-12(抗pd-1肽)。
[0202]
在一些实施方案中,免疫检查点分子的抑制剂为pd-l1的抑制剂,例如,抗pd-l1单克隆抗体。在一些实施方案中,抗pd-l1单克隆抗体为bms-935559、bms-936559(mdx-1105)、medi-4736(德瓦鲁单抗(durvalumab))、mpdl3280a(亦称为rg7446)、yw243.55.s70(hpab-0381-wj)、或msb0010718c。在一些实施方案中,抗pd-l1单克隆抗体为mpdl3280a或medi-4736。某些实施方案中,抗pd-l1抗体包括阿特珠单抗(atezolizumab)、阿维鲁单抗(avelumab)、德瓦鲁单抗(durvalumab)或medi-4736,及mpdl3280a。
[0203]
在一些实施方案中免疫检查点分子的抑制剂为ctla-4的抑制剂,例如,抗ctla-4抗体。在一些实施方案中,抗ctla-4抗体为伊匹单抗(ipilimumab)、曲美木单抗(tremelimumab),或wo2014/207063所揭示的任何抗体(已以引用的方式并入本文中)。其他分子包括多肽,或可溶性突变体cd86多肽。某些实施方案中,抗体为伊匹单抗(ipilumumab)。
[0204]
某些实施方案中,免疫检查点分子的抑制剂为两种或更多种本文所描述调控剂的组合,如靶向两种或更多种不同靶标的组合(例如,pd-1、pd-l1及pd-l2)。示例性组合包括:α-pd-1与α-pd-l1;α-ctla-4、α-pd-l1、与α-cd20等等。
[0205]
在一些实施方案中,免疫检查点分子的抑制剂为阻断或抑制4-1bb与其配体间相互作用的抗体,包括乌特鲁单抗(utomilumab)。
[0206]
在一些实施方案中,免疫检查点分子的抑制剂为csfir的抑制剂,例如,抗csf1r抗体。在一些实施方案中,抗csf1r抗体为imc-cs4或rg7155。
[0207]
在一些实施方案中,免疫检查点分子的抑制剂为lag3的抑制剂,例如,抗lag3抗体。在一些实施方案中,抗lag3抗体为bms-986016、lag525、imp321或gsk2831781。
[0208]
在一些实施方案中,免疫检查点分子的抑制剂为gitr的抑制剂,例如,抗gitr抗体。在一些实施方案中,抗gitr抗体为trx518、mk-4166、mk1248、bms-986156、medi1873、或gwn323。
[0209]
在一些实施方案中,免疫检查点分子的抑制剂为ox40的抑制剂,例如,抗ox40抗体或ox40l融合蛋白质。在一些实施方案中,抗ox40抗体为medi0562、medi6469、moxr0916、pf-04518600、或gsk3174998。在一些实施方案中,ox40l融合蛋白质为medi6383。
[0210]
在一些实施方案中,免疫检查点分子的抑制剂为tim3的抑制剂,例如,抗tim3抗体。在一些实施方案中,抗tim3抗体为mbg-453。
[0211]
在一些实施方案中,免疫检查点分子的抑制剂为cd20的抑制剂,例如,抗cd20抗体。在一些实施方案中,抗cd20抗体为奥宾珠单抗(obinutuzumab)或利妥昔单抗(rituximab)。
[0212]
在一些实施方案中,本发明的化合物可以与一种或多种代谢酶抑制剂联合使用。在一些实施方案中,代谢酶抑制剂为ido l、tdo、或精氨酸酶的抑制剂。ido1抑制剂的实例包括艾卡哚司他(epacadostat)及ngl919。精氨酸酶抑制剂的实例为cb-1158。
[0213]
在一些实施方案中,本发明的化合物可以与双特异性抗体联合使用。在一些实施方案中,双特异性抗体的其中一个域靶向pd-1、pd-l1、ctla-4、gitr、ox40、tim3、lag3、cd137、icos、cd3或tgfβ受体。
[0214]
在一些实施方案中,本发明的化合物可以与用于治疗疾病(如癌症)的一种或多种药剂联合使用。在一些实施方案中,该药剂为烷化剂、蛋白酶体抑制剂、皮质类固醇或免疫调控剂。烷化剂实例包括苯达莫司汀(bendamustine)、氮芥(nitrogen mustard)、乙烯亚胺衍生物、磺酸烷基酯、硝基脲与三氮烯类、尿嘧啶芥(uracil mustard)、氮芥(chlormethine)、环磷酰胺(cytoxan
tm
)、异环磷酰胺(ifosfamide)、马法兰(melphalan)、苯丁酸氮芥(chlorambucil)、双溴丙基呱嗪(pipobroman)、三乙烯-密胺(triethylene-melamine)、三亚乙基硫代磷酰胺(triethylenethiophosphoramine)、白消安(busulfan)、卡莫司汀(carmustine)、洛莫司汀(lomustine)、链脲佐菌素(streptozocin)、达卡巴仁(dacarbazine)和替莫唑胺(temozolomide)。在一些实施方案中,蛋白酶体抑制剂为卡非佐米(carfilzomib)。在一些实施方案中,皮质类固醇为地塞美松(dexamethasone)(dex)。在一些实施方案中,免疫调控剂为来那度胺(lenalidomide)(len)或泊马度胺(pomalidomide)(pom)。
[0215]
本发明的化合物可以进一步与其他治疗癌症的方法联合使用,例如,化疗法、放射疗法、肿瘤靶向疗法、化疗前置疗法、免疫疗法或手术。免疫疗法实例包括细胞因子治疗法(例如,干扰素、gm-csf、g-csf、il-2)、crs-207免疫疗法、癌症疫苗、单克隆抗体、过继性t细胞转移、溶瘤病毒疗法及免疫调控小分子,包括沙利度胺(thalidomide)或jak1/2抑制剂及类似物。
[0216]
本发明的化合物可以组合施用一多种抗癌药物,如化疗剂。化疗法实例包括以下任何:阿巴瑞克(abarelix)、阿比特龙(abiraterone)、阿法替尼(afatinib)、阿柏西普(aflibercept)、阿地白介素(aldesleukin)、阿仑单抗(alemtuzumab)、阿利维甲酸(alitretinoin)、别嘌呤醇(allopurinol)、六甲蜜胺(altretamine)、安耐曲唑(anastrozole)、三氧化二砷、天冬酰胺酶、阿西替尼(axitinib)、阿扎胞苷(azacitidine)、贝伐单抗(bevacizumab)、贝瑟罗汀(bexarotene)、巴瑞替尼(baricitinib)、比卡鲁胺(bicalutamide)、博来霉素(bleomycin)、硼替佐必(bortezombi)、硼替佐米(bortezomib)、布立尼布(brivanib)、布帕尼西(buparlisib)、白消安(busulfan)(静脉内)、白消安(口
服)、二甲睾酮(calusterone)、卡培他滨(capecitabine)、卡铂(carboplatin)、卡莫司汀(carmustine)、西地尼布(cediranib)、西妥昔单抗(cetuximab)、苯丁酸氮芥(chlorambucil)、顺铂(cisplatin)、克拉屈滨(cladribine)、克罗拉滨(clofarabine)、克唑替尼(crizotinib)、环磷酰胺、阿糖胞苷(cytarabine)、达卡巴仁(dacarbazine)、达克替尼(dacomitinib)、更生霉素(dactinomycin)、达肝素钠(dalteparin sodium)、达沙替尼(dasatinib)、更生霉素(dactinomycin)、唐霉素(daunorubicin)、地西他滨(decitabine)、地加瑞克(degarelix)、地尼白介素(denileukin)、地尼白介素迪夫托斯(denileukin diftitox)、脱氧柯福霉素(deoxycoformycin)、右雷佐生(dexrazoxane)、欧洲紫杉醇(docetaxel)、多柔比星(doxorubicin)、卓罗沙吩(droloxafine)、丙酸屈他雄酮(dromostanolone propionate)、抑克珠单抗(eculizumab)、恩杂鲁胺(enzalutamide)、表足叶草毒(epidophyllotoxin)、泛艾霉素(epirubicin)、厄洛替尼(erlotinib)、雌氮芥(estramustine)、磷酸依托泊苷(etoposide phosphate)、依托泊苷(etoposide)、依西美坦(exemestane)、柠檬酸芬太尼(fentanyl citrate)、非格司亭(filgrastim)、氟尿嘧啶脱氧核苷(floxuridine)、福达乐(fludarabine)、氟尿嘧啶(fluorouracil)、氟他胺(flutamide)、氟维司群(fulvestrant)、吉非替尼(gefitinib)、吉西他滨(gemcitabine)、吉妥珠单抗(gemtuzumab ozogamicin)、乙酸戈舍瑞林(goserelin acetate)、乙酸组胺瑞林(histrelin acetate)、替伊莫单抗(ibritumomab tiuxetan)、伊达比星(idarubicin)、艾德拉尼(idelalisib)、异环磷酰胺(ifosfamide)、甲磺酸伊马替尼(imatinib mesylate)、干扰素α2a、伊立替康(irinotecan)、二甲苯磺酸拉帕替尼(lapatinib ditosylate)、来那度胺(lenalidomide)、来曲唑(letrozole)、若克瘤(leucovorin)、乙酸柳菩林(leuprolide acetate)、左旋咪唑(levamisole)、洛莫司汀(lomustine)、甲氮芥(meclorethamine)、乙酸甲地孕酮(megestrol acetate)、马法兰(melphalan)、巯基嘌呤、甲胺蝶呤(methotrexate)、甲氧沙林(methoxsalen)、光辉霉素(mithramycin)、丝裂霉素(mitomycin c)、米托坦(mitotane)、米托蒽醌(mitoxantrone)、苯丙酸诺龙(nandrolone phenpropionate)、温诺平(navelbene)、耐昔妥珠单抗(necitumumab)、奈拉滨(nelarabine)、来那替尼(neratinib)、尼洛替尼(nilotinib)、尼鲁米特(nilutamide)、若莫单抗(nofetumomab)、欧色林(oserelin)、奥沙利铂(oxaliplatin)、太平洋紫杉醇(paclitaxel)、裴米卓耐特(pamidronate)、帕尼单抗(panitumumab)、帕唑帕尼(pazopanib)、培门冬酶(pegaspargase)、培非格司亭(pegfilgrastim)、培美曲塞二钠(pemetrexed disodium)、喷司他汀(pentostatin)、皮拉利希(pilaralisib)、双溴丙基呱嗪(pipobroman)、普卡霉素(plicamycin)、普纳替尼(ponatinib)、泼尼松(prednisone)、丙卡巴肼(procarbazine)、奎纳克林(quinacrine)、拉布立酶(rasburicase)、瑞格非尼(regorafenib)、瑞罗沙吩(reloxafine)、利妥昔单抗(rituximab)、鲁索利替尼(ruxolitinib)、索拉非尼(sorafenib)、链脲佐菌素(streptozocin)、舒尼替尼(sunitinib)、马来酸舒尼替尼、泰莫西芬(tamoxifen)、替加氟(tegafur)、替莫唑胺(temozolomide)、替尼泊苷(teniposide)、睾内酯(testolactone)、沙利度胺(thalidomide)、硫鸟嘌呤(thioguanine)、噻替呱(thiotepa)、托普替康(topotecan)、托瑞米芬(toremifene)、托西莫单抗(tositumomab)、曲妥珠单抗(trastuzumab)、维甲酸(tretinoin)、曲普瑞林(triptorelin)、尿嘧啶氮芥、戊柔比星(valrubicin)、凡德他尼
(vandetanib)、长春花碱(vinblastine)、长春新碱(vincristine)、长春地辛(vindesine)、长春瑞滨(vinorelbine)、伏立诺他(vorinostat)及唑来膦酸(zoledronate)。
[0217]
其他抗癌剂包括抗体疗法,如曲妥珠单抗(trastuzumab)(赫塞汀(herceptin))、针对共刺激分子的抗体如ctla-4(例如,伊匹单抗(ipilimumab)或曲美木单抗(tremelimumab))、4-1bb、针对pd-1及pd-l1的抗体、或针对细胞因子(il-10、tgf-β等等)的抗体。可与本发明的化合物联合用于治疗癌症或感染(如病毒、细菌、真菌和寄生物感染)的针对pd-1和/或pd-l1的抗体实例包括(但不限于)纳武单抗(nivolumab)、帕博利珠单抗(pembrolizumab)、mpdl3280a、medi-4736和shr-1210。
[0218]
其他抗癌剂包括与细胞增殖性病症相关的激酶的抑制剂。这些激酶包括但不限于aurora-a、cdk1、cdk2、cdk3、cdk5、cdk7、cdk8、cdk9、ephrin受体激酶、chk1、chk2、src、yes、fyn、lck、fer、fes、syk、itk、bmx、gsk3、jnk、pakl、pak2、pak3、pak4、pdkl、pka、pkc、rsk及sgk。
[0219]
其他抗癌剂亦包括阻断免疫细胞迁移的那些,如趋化因子受体(包括ccr2及ccr4)的拮抗剂。本公开的化合物可进一步与一种或多种抗炎剂、类固醇、免疫抑制剂或治疗性抗体联合使用。
[0220]
在一些实施方案中,本发明的化合物可以与直接刺激免疫效应子反应的其他治疗剂,如细胞因子,或肿瘤特异性过继转移t细胞群,或特异于肿瘤细胞所表达蛋白质的抗体联合使用。
[0221]
如本文所用,“直接刺激免疫效应子反应的药剂”意指任何合适药剂,但通常指细胞因子或趋化因子(或刺激产生其中一种的药剂)、肿瘤特异性过继转移t细胞群,或特异于肿瘤细胞所表达蛋白质的抗体。
[0222]
细胞因子可为选自以下的干扰素:ifnα、ιρνβ、ifnγ及ifna、或白介素,如il-2。趋化因子可能为炎性介质,例如,选自cxcl9、10和11,其吸引表达cxcr3的t细胞。该刺激产生细胞因子或趋化因子的药剂可能为适合施用给人的佐剂。一种实例为卡介苗(bacille calmette-guerin)(bcg),其通常施用至膀胱内(亦即尿道),用于治疗膀胱癌。bcg用于膀胱癌的典型剂量方案为每周一次共6周,但为了长期安全性病史,亦可无限期维持施用。已知bcg可刺激对膀胱癌的免疫应答。bcg亦已用为佐剂,与包含肿瘤抗原的组合物(亦即使用癌症疫苗)联合,通常当皮内施用时,特别用于结肠癌。此类bcg用法亦涵盖在本发明内。肿瘤特异性过继性转移t细胞群直接增加受试者中肿瘤特异性t细胞群数量,且可采用任何合适方式产生。然而,该过程通常涉及从来自患者的肿瘤样品中分离肿瘤特异性t细胞,并选择性培养那些细胞后,再将已扩增的肿瘤特异性t细胞群送回患者。或者,可采用遗传工程处理t细胞受体基因座,然后扩增已改变的细胞,产生肿瘤特异性t细胞群。
[0223]
特异性针对肿瘤细胞所表达蛋白质的抗体通常通过与肿瘤细胞结合来刺激免疫活性,并经由抗体依赖性细胞介导细胞毒性(adcc)促进细胞瓦解。此类抗体实例包括抗cd20抗体,如奥法木单抗(ofatumumab)或利妥昔单抗(rituximab),及抗cd52抗体,如阿仑单抗(alemtuzumab)。
[0224]
因此某些示例性实施方案中,本发明的化合物可以与钙调磷酸酶(calcineurin)抑制剂联合使用,例如,环孢素a或fk 506;mtor抑制剂,例如,雷帕霉素(rapamycin)、40-0-(2-羟基乙基)-雷帕霉素(rapamycin)、优美莫司(biolimus)-7或优美莫司(biolimus)-9;
具有免疫抑制性质的子囊霉素(ascomycin),例如,abt-281、asm981;皮质类固醇;环磷酰胺;硫唑嘌呤(azathioprene);甲氨蝶呤(methotrexate);来氟米特(leflunomide);咪唑立宾(mizoribine);霉酚酸或盐;霉酚酸莫太盐(mycophenolate mofetil);il-1β抑制剂。
[0225]
另一个实施方案中,本发明的化合物与为pi3激酶抑制剂的并用剂组合。
[0226]
另一个实施方案中,本发明的化合物与影响btk(布鲁顿氏(bruton’s)酪氨酸激酶)的并用剂组合。
[0227]
用于治疗肿瘤疾病的本发明的化合物可以与b-细胞调控剂联合使用,例如,利妥昔单抗(rituximab)、btk或syk抑制剂;pkc、pi3激酶、pdk、pim、jak与mtor及bh3模拟物的抑制剂。
[0228]
在一些实施方案中,本发明的化合物(包括其盐)可以与另一种免疫原剂组合,如癌细胞、纯化的肿瘤抗原(包括重组蛋白质、肽和碳水化合物分子)、细胞、以及经过编码免疫刺激细胞因子的基因转染的细胞。可使用的肿瘤疫苗的无限制实例包括黑色素瘤抗原的肽,如gp100的肽、mage抗原、trp-2、marti和/或酪氨酸酶,或已转染而表达细胞因子gm-csf的肿瘤细胞。
[0229]
在一些实施方案中,本发明的化合物或其盐亦可用于与治疗癌症的疫苗接种过程组合。在一些实施方案中,该肿瘤细胞经过转导而表达gm-csf。在一些实施方案中,肿瘤疫苗包括来自涉及人癌症的病毒的蛋白质,如人乳突病毒(hpv)、肝炎病毒(hbv及hcv)及卡波西疱疹肉瘤病毒(khsv)。在一些实施方案中,本发明的化合物可以与肿瘤特异性抗原,如从肿瘤组织本身分离的热休克蛋白联合使用。在一些实施方案中,本发明的化合物或其盐可以与树突状细胞免疫接种法组合,以激活强力的抗肿瘤应答。
[0230]
在一些实施方案中,本发明的化合物可以联合双特异性大环肽使用,该双特异性大环肽使表达feα或feγ受体的效应子细胞靶向肿瘤细胞。本发明的化合物亦可以与激活宿主免疫应答的大环肽组合。
[0231]
在一些实施方案中,本发明的化合物可以联合骨髓移植物使用,用于治疗各种不同造血来源的肿瘤。
[0232]
旨在与本发明的化合物联合使用的合适抗病毒剂包含核苷及核苷酸逆转录酶抑制剂(nrti)、非核苷逆转录酶抑制剂(nnrti)、蛋白酶抑制剂及其他抗病毒药物。合适nrti的实例包括齐多夫定(zidovudine)(azt);地达诺新(didanosine)(ddl);扎西他滨(zalcitabine)(ddc);司他夫定(stavudine)(d4t);拉米夫定(lamivudine)(3tc);阿巴卡维(abacavir)(1592u89);阿德福韦酯(adefovir dipivoxil)[双(pom)-pmea];洛布卡韦(lobucavir)(bms-180194);bch-10652;恩曲他滨(emitricitabine)[(-)-ftc];β-l-fd4(亦称为β-l-d4c,命名为β-l-2’,3
’‑
二去氧-5-氟-胞苷);dapd((-)-β-d-2,6-二氨基-嘌呤二氧杂环戊烷);及二去氧腺苷(lodenosine)(fdda)。典型的合适nnrti包括奈韦拉平(nevirapine)(bi-rg-587);地拉韦啶(delaviradine)(bhap、u-90152);依法韦仑(efavirenz)(dmp-266);pnu-142721;ag-1549;mkc-442(l-(乙氧基-甲基)-5-(l-甲基乙基)-6-(苯基甲基)-(2,4(1h,3h)-嘧啶二酮);及(+)-胡桐素(calanolide)a(nsc-675451)及b。典型的合适蛋白酶抑制剂包括沙奎那维(saquinavir)(ro 31-8959);利托那韦(ritonavir)(abt-538);茚地那韦(indinavir)(mk-639);奈非那韦(nelfnavir)(ag-1343);安佩那韦(amprenavir)(141w94);拉西那韦(lasinavir)(bms-234475);dmp-450;
bms-2322623;abt-378;及ag-1549。其他抗病毒剂包括羟基脲、利巴韦林(ribavirin)、il-2、il-12、喷他夫西(pentafuside)和yissum project no.11607。
[0233]
据了解,本发明方法所使用许多另一种治疗剂可能为需要经静脉内、腹膜内,或储积式施用的生物制剂。在又一个实施方案中,本发明的化合物经口施用,而另一种治疗剂则胃肠外施用,例如,经静脉内、经腹膜内或呈储积物施用。
[0234]
本文描述的任何组合疗法中,当对患者施用超过一种药物制剂时,其可同时、分开、序贯或组合施用(例如,超过两种药剂时)。
[0235]
在一个实施方案中,本发明提供了包含本发明的化合物的产品(如主要化合物或其任何亚家族)及至少一种其他治疗剂,成为组合制剂,用于同时、分开、或序贯用于疗法。呈组合制剂提供的产品包括在同一个药物组合物中包含本发明的化合物或其任何亚家族及其他治疗剂(群)的组合物,或呈分开形式包含主要化合物或其任何亚家族及其他治疗剂(群)的组合物,例如,呈试剂盒型式。
[0236]
在一个实施方案中,本发明提供了包含两种或更多种单独药物组合物的试剂盒,其中至少一种包含主要化合物,及另一种包含本文所讨论的第二治疗剂。在一个实施方案中,试剂盒包含用于分开收纳所述组合物的手段,如容器、分开的瓶子,或分开的箔包。此类试剂盒的实例为泡壳包,通常用于包装片剂、胶囊及类似物。本发明试剂盒可用于在不同施用间隔施用不同剂型,例如,经口及胃肠外,施用分开的组合物,或由分开的组合物彼此调整剂量。为了促进适应性,本发明试剂盒通常包含施用说明书。
[0237]
6.化合物筛选/测定方法
[0238]
本发明的化合物抑制hpk1的激酶活性,该激酶活性可直接采用许多种生化测定法来测定,如实施例1中描述的测定法。可根据一个抑制剂浓度范围来确定任何化合物的ic50值。此外,化合物的抑制作用亦可采用生物测定法评估,以确定化合物对t细胞受到tcr及cd28刺激后的细胞因子分泌的影响。
[0239]
例如,实施例5描述此类功能测定法,用于决定hpk1抑制剂对受到刺激的pan t细胞的il-2&ifn-γ释放的影响。可采用标准elisa测定法测量/定量所分泌的il-2&ifn-γ。简言之,采用自商品购得的试剂盒,如:macs(miltenyl biotec)pan t分离试剂盒(目录号130-096-535),自外周血(pb)单个核细胞(mnc)、或pbmc分离出pan t细胞。原代人pan-t细胞包括cd4与cd8t细胞及一些γ/δt细胞亚群。pan-t细胞可以使用阴性免疫磁性分离技术分离,不使用柱。
[0240]
分离的pan-t细胞以100,000个细胞/孔分配在96孔板中,使用固定化抗cd3抗体及可溶性抗cd28抗体刺激,或使用pma/离子霉素(ionomycin)作为阳性对照(或作为阴性对照的培养基)。可添加不同浓度的测试化合物至细胞中,在受到tcr-刺激/cd28共同刺激后,评估化合物对细胞因子分泌的影响。受刺激的细胞再温育2天后,收集各孔的上清液(包含pan-t细胞分泌的细胞因子),供进行elisa测定法,及定量il-2及ifn-γ。
[0241]
亦可采用其他测定法来评估任何hpk1抑制剂抑制hpk1的能力,或筛选具有hpk1抑制活性的化合物。
[0242]
例如,在一种测定法中,可采用下文描述的treg测定法(调节性t-细胞增殖测定法)来测定对hpk1激酶活性的抑制性。明确言之,采用合适试剂盒,如一种来自thermo fisher scientific(目录号11363d)的试剂盒,从人体捐赠的外周血单个核细胞(pbmc)中
分离原代cd4
+
/cd25-t-细胞及cd4
+
/cd25
+
调节性t-细胞。根据供应商提供的程序,在cd4
+
/cd25-t-细胞上标记cfse(thermo fisher scientific,c34554)。将标记cfse的t-细胞及cd4
+
/cd25
+
调节性t-细胞以1
×
106个细胞/ml的浓度再悬浮于rpmi-1640培养基中。将100μl标记cfse的t-细胞与或不与50μl cd4
+
/cd25
+
调节性t-细胞混合,使用5μl抗cd3/cd28珠(thermo fisher scientific,11132d)及于50μl rpmi-1640培养基中稀释成各种不同浓度的化合物处理。混合的细胞群培养5天(37℃,5%co2),于第5天时,采用bd lsrfortessa x-20,使用fitc途径,分析标记cfse的t-细胞的增殖。预期该主要化合物抑制hpk1,以增强treg功能及抑制标记cfse的原代cd4
+
/cd25-t-细胞的增殖。
[0243]
另一实例中,可以采用下文描述的p-slp-76s376htrf测定法(cisbio)测定对hpk1激酶活性的抑制性。此基于htrf细胞的测定法可以快速定量检测通过hpk1的slp-76在丝氨酸376的磷酸化。磷-slp-76成为一个可在上面建立关键性信号传导复合物的支架,并作为t-淋巴细胞活化作用的标志物。根据制造商,磷-slp-76(ser376)测定法使用两种标记抗体:一种具有供体荧光团,另一种具有接受体。第一种抗体特异性针对slp-76上磷酸化s376基序的结合,及第二种是针对其识别不依赖其磷酸化状态的slp-76的能力。蛋白质磷酸化可以形成涉及两种标记抗体的免疫-复合物,并将供体荧光团带到紧邻接受体的附近,藉以产生fret信号。其强度与样品中所含磷酸化蛋白质浓度成正相关性,并提供一种在免洗测定模式(no-wash assay format)下评估蛋白质磷酸化状态的方式。
[0244]
简言之,收集jurkat细胞(于含10%fbs的rpmi1640培养基中培养)及离心后,再以3
×
106个细胞/ml悬浮于适当培养基中。然后将jurkat细胞(35μl)分配至384孔板的各孔中。用细胞培养基稀释测试化合物,以获得40倍稀释物(添加39μl细胞培养基至1μl化合物中)。使用各种不同浓度的测试化合物(添加5ml稀释化合物至35μl jurkat细胞中,从3μμ开始稀释1:3)处理多孔板中的jurkat细胞,于37℃及5%co2进行1小时,然后使用抗cd3(5μg/ml,okt3纯系)处理30min,以激活tcr及hpk1。使用4
×
裂解缓冲液(lb)制备100
×
封闭试剂(来自p-slp76ser376htrf试剂盒)的1:25稀释物,并将15μl含封闭试剂的4
×
lb缓冲液添加至各孔中,并在室温下温和摇动温育45mins。将细胞裂解物(16μl)加至greiner白色板中,使用p-slp76ser376htrf试剂(2μl供体,2μl接受体)处理,及于4℃温育过夜。次日,在pherastar读板器上测定均相时差性荧光(htrf)。采用graphpad prism 5.0软件,由抑制百分比拟合相对于抑制剂浓度的曲线,确定ic50。
[0245]
上述测定法中的任一者均可以放大规模或放大高通量筛选(hts)。可采用上述测定法中的任一者确定主要化合物的ic50。
[0246]
7.药物组合物
[0247]
本发明提供了药物组合物,其包含本文所述化合物中的任一者、或其药学上可接受的盐,及一种或多种药学上可接受的载剂或赋形剂。
[0248]“药学上可接受的赋形剂”及“药学上可接受的载剂”是指协助调配和/或施用活性剂给受试者和/或被受试者吸收的物质,并可包括在本发明组合物中,不会对受试者引起显著的不良毒性副作用。药学上可接受的载剂及赋形剂的非限制实例包括水、nacl、生理盐水溶液、乳酸化林格氏(ringer’s)溶液、生理蔗糖溶液、生理葡萄糖溶液、结合剂、填料、崩解剂、润滑剂、包衣剂、甜味剂、调味剂、盐溶液(如林格氏溶液)、醇类、油类、明胶、碳水化合物(如乳糖、直链淀粉或淀粉)、脂肪酸酯类、羟基甲基纤维素、聚乙烯吡咯啶,与着色剂及类似
物。此类制剂可经过杀菌,若需要时,再与辅剂混合,所述辅剂不与本文所提供化合物的活性有不良反应或干扰性,如润滑剂、防腐剂、安定剂、湿化剂、乳化剂、影响渗透压的盐、缓冲剂、着色剂和/或芳香物质及类似物。本领域技术人员将了解其他适合本发明的化合物使用的药学上的载剂及赋形剂。
[0249]
这些组合物任选地进一步包含一种或多种额外治疗剂。或者,本发明的化合物可以向有此需要的患者组合施用一种或多种其他治疗性方案(例如,gleevec或其他激酶抑制剂、干扰素、骨髓移植、法呢基(farnesyl)转移酶抑制剂、双磷酸盐类、沙利度胺(thalidomide)、癌症疫苗、激素疗法、抗体、放射线等等)。例如,用于与本发明的化合物共同施用或包含在药物组合物中的额外治疗剂可能为另一种或多种抗癌剂。
[0250]
如本文所述,本发明组合物包含本发明的化合物及药学上可接受的载剂,如本文所用,其包括适合期望特定剂型的任何及所有溶剂、稀释剂、或其他媒介物、匀散剂或悬浮助剂、界面活性剂、等渗剂、增稠剂或乳化剂、防腐剂、固体结合剂、润滑剂及类似物。remington’s pharmaceutical sciences,第十五版,e.w.martin(mack publishing co.,easton,pa.,1975)揭示用于调配药物组合物的各种不同载剂及其制法的已知技术。除非任何公知载剂介质无法与本发明的化合物相容,如会产生任何不期望的生物效应或会以有害方式与药物组合物中任何其他组分(群)相互作用,否则其用法均旨在涵盖在本发明范围内。有些可作为药学上可接受的载剂的材料实例包括但不限于:糖类(如乳糖、葡萄糖及蔗糖);淀粉(如玉米淀粉及马铃薯淀粉);纤维素及其衍生物(如羧基甲基纤维素钠、乙基纤维素及纤维素乙酸酯);黄芪胶粉末;麦芽;明胶;滑石;赋形剂(如可可奶油及栓剂蜡);油类,如花生油、棉籽油、葵花油、芝麻油、橄榄油、玉米油及大豆油;二醇类,如丙二醇;酯类,如油酸乙酯及月桂酸乙酯;琼脂;缓冲剂,如氢氧化镁及氢氧化铝;藻酸;无热原水;等渗性等渗盐水;林格氏溶液;乙醇,及磷酸盐缓冲溶液,以及其他无毒的相容性润滑剂,如月桂基硫酸钠及硬脂酸镁,以及着色剂、释放剂、包衣剂、甜味剂、调味剂及香料剂、防腐剂及抗氧化剂亦可含在组合物中。
[0251]
8.配制剂
[0252]
本发明亦包括一类别组合物,其包含本发明活性化合物与一种或多种药学上可接受的载剂和/或稀释剂和/或佐剂(本文中统称为“载剂”材料),并且若期望时,与其他活性成分缔合。
[0253]
某些实施方案中,本发明提供了用于治疗癌症,特别是本文所述癌症的药物配制剂,其包含本发明的化合物或其药学上可接受的盐,及药学上可接受的载剂。
[0254]
某些实施方案中,本发明提供了治疗选自下列各物所组成群中的癌症的药物配制剂:乳腺癌、结直肠癌、肺癌、卵巢癌和胰腺癌,其包含本发明的化合物或其药学上可接受的盐,与药学上可接受的载剂。
[0255]
本发明的化合物可采用任何合适途径施用,优选为呈配合此类途径的药物组合物形式及可有效治疗的剂量施用。本发明的化合物及组合物例如,可以经口、粘膜、表面、直肠、肺(如吸入式喷雾)或胃肠外(包括经血管、静脉内、腹膜内、皮下、肌内、穿皮、眼框内、鞘内、脑室内、肿瘤内、鼻内、颅内、植入、吸入、以及采用输注技术,呈包含药学上可接受的载剂、佐剂和媒介物的剂量单位配制剂施用。
[0256]
典型地,药物组合物为片剂或明胶胶囊,其包含活性成分,连同a)稀释剂,例如,乳
糖、右旋糖、蔗糖、甘露糖醇、山梨糖醇、纤维素和/或甘氨酸;b)润滑剂,例如,硅石、滑石、硬脂酸、其镁盐或钙盐和/或聚乙二醇;用于片剂还有c)结合剂,例如,硅酸镁铝、淀粉糊、明胶、黄芪胶、甲基纤维素、羧甲基纤维素钠和/或聚乙烯吡咯烷酮;若期望时,d)崩解剂,例如,淀粉、琼脂、藻酸或其钠盐,或泡腾混合物;和/或e)吸收剂、着色剂、调味剂和甜味剂。片剂可以根据本领域已知的技术包被膜衣或肠衣。
[0257]
用于口服施用的合适组合物包括有效量的本发明的化合物,其呈片剂、锭剂、水性或油性悬浮液、分散性粉剂或粒剂、乳液、硬胶囊或软胶囊、或糖浆或酏剂的形式。旨在口服使用的组合物是根据本领域已知用于制造药物组合物的任何方法制备,并且此类组合物可以含有选自由以下组成的组一种或多种药剂:甜味剂、调味剂、着色剂及防腐剂,以提供药学上美观且适口的制剂。片剂可以含有与适合制造片剂的无毒药学上可接受的赋形剂混合的活性成分。此类赋形剂为例如,惰性稀释剂,如碳酸钙、碳酸钠、乳糖、磷酸钙或磷酸钠;造粒剂及崩解剂,例如,玉米淀粉,或藻酸;结合剂,例如,淀粉、明胶或阿拉伯树胶;及润滑剂,例如,硬脂酸镁、硬脂酸或滑石。片剂可以未包被,或通过已知技术包被,以便在胃肠道中延缓崩解及吸收,从而在较长时期内提供持续作用。可使用例如,延时材料,如单硬脂酸甘油酯或二硬脂酸甘油酯。用于口服使用的配制剂可以以硬明胶胶囊呈现,其中活性成分与惰性固体稀释剂(例如,碳酸钙、磷酸钙或高岭土)混合,或以软明胶胶囊呈现,其中活性成分与水或油介质(例如,花生油、液态石蜡或橄榄油)混合。
[0258]
某些可注射组合物为水性等渗溶液或悬浮液,并且栓剂有利地由脂肪乳液或悬浮液制备。所述组合物可以经过杀菌和/或包含佐剂,如防腐剂、安定剂、湿化剂或乳化剂、促进溶解剂、调整渗透压的盐和/或缓冲剂。此外,其亦可包含其他具有治疗价值的物质。所述组合物分别根据常规混合、造粒或包衣方法制备,并且含有约0.1-75%或含有约1-50%的活性成分。用于经皮应用的合适组合物包括有效量的本发明的化合物及合适的载剂。适用于经皮递送的载剂包括可吸收的药学上可接受的溶剂,以促进通过宿主皮肤。例如,经皮装置呈绷带形式,其包含背衬元件、含有化合物及任选地具有载剂的贮液器、任选地速率控制障壁,以便以控制且预定的速率长期递送化合物给宿主皮肤,及包含可固定该装置在皮肤上的方式。
[0259]
用于表面应用,例如至皮肤及眼睛的合适组合物,包括水性溶液、悬浮液、软膏剂、乳霜、凝胶或可喷洒配制剂,例如,采用气溶胶或类似物递送。此类表面递送系统将特别适合皮肤应用,例如用于皮肤癌症的治疗,例如,用于在防晒乳、洗液、喷雾及类似物中的防治性用途。因此其特别适用于表面,包括本领域公知的配制剂,化妆品。其可以含有溶解剂、安定剂、张力增强剂、缓冲剂及防腐剂。
[0260]
如本文所用,表面应用还可以适合吸入或鼻内应用。其可以方便地以干粉(单独、或作为混合物,例如与乳糖的干混物,或混合组分颗粒,与例如与磷脂形成)的形式从干粉吸入器递送,或使用或不使用合适推进剂下,呈气溶胶喷雾从加压容器、泵、喷瓶、雾化器或喷雾器递送。
[0261]
本发明进一步提供包含本发明的化合物作为活性成分的无水药物组合物及剂型,因为水可能促使某些化合物的降解。
[0262]
本发明的无水药物组合物及剂型可以使用无水或低水分的成分和在低水分及低湿度条件下制备。可以制备及储存无水药物组合物,因此维持其无水性质。因此,使用已知
可以阻止暴露于水的材料包装无水组合物,以便其可容纳在合适的配制盒内。合适包装的实例包括但不限于密封箔、塑料、单位剂量容器(例如,小瓶)、罩板包装及长条包装。
[0263]
本发明进一步提供了药物组合物及剂型,其包含可以降低作为活性成分的本发明的化合物的降解速率的一种或多种药剂。此类药剂在本文中称为“安定剂”,包括但不限于抗氧化剂,如抗坏血酸、ph缓冲剂或盐缓冲剂等等。
[0264]
本发明药物活性化合物可根据制药上公知的方法制成施用于患者(包括人及其他哺乳动物)的药剂。
[0265]
使用根据本发明的化合物和/或组合物治疗疾病状况的化合物施用量及剂量方案依据各种不同因素而定,包括受试者的年龄、体重、性别及医学状况、疾病类型、疾病严重性、施用途径与频率及所采用的特定化合物。因此,剂量方案可能有很大变化,但照例由标准方法决定。如前述,日剂量可以施用一次或可分成2、3、4或更多次施用。
[0266]
基于治疗目的,本发明活性化合物通常与适合指定施用途径的一种或多种佐剂、赋形剂或载剂组合。若经口施用时,化合物可与乳糖、蔗糖、淀粉粉末、烷酸类的纤维素酯类、纤维素烷基酯、滑石、硬脂酸、硬脂酸镁、氧化镁、磷酸或硫酸的钠盐及钙盐、明胶、阿拉伯树胶、藻酸钠、聚乙烯吡咯烷酮、和/或聚乙烯醇混合,及接着压片或囊封,以方便施用。此类胶囊或片剂可包含控制释放配制剂,因为可以让活性化合物匀散在羟基丙基甲基纤维素中。
[0267]
以皮肤状况为例,其优选是在受影响区域施加本发明的化合物的表面制剂,一天2至4次。适合表面施用的配制剂包括适合渗透皮肤的液体或半液体制剂(例如,擦剂、洗液、软膏剂、乳霜、或糊剂),及适合施用至眼睛、耳朵或鼻的滴剂。供表面施用的活性成分可占配制剂的0.001%至10%w/w,例如,1%至2%重量比,但其可能占配制剂高达10%w/w,但优选不超过5%w/w,及更优选地为0.1%至1%。
[0268]
本发明的化合物亦可采用经皮装置施用。优选地利用贴布达成经皮施用,贴布呈贮液器与多孔膜型或呈固体基质变化型。这两种情况下的活性剂均从贮液器或微胶囊通过膜连续递送至与接受者的皮肤或粘膜接触的活性剂通透性胶粘剂中。若穿过皮肤吸收活性剂,则以控制且预定的流速施用活性剂给接受者。以微胶囊为例,囊封剂亦具有作为膜的功能。本发明乳液的油相可由已知成分,以已知方式构成。
[0269]
虽然该相可仅包含乳化剂,但其可能包含至少一种乳化剂与脂肪或油或脂肪与油二者的混合物。优选地,包括亲水性乳化剂与亲脂性乳化剂共同作为安定剂。亦优选是同时包括油与脂肪。总言之,乳化剂(群)使用或不使用安定剂(群)构成所谓的乳化蜡,及该蜡连同油与脂肪共同构成所谓的乳化软膏剂基质,形成乳霜配制剂的油匀散相。适用于本发明的配制剂的本发明乳化剂及乳液安定剂包括tween 60、span 80、鲸蜡硬脂醇、肉豆蔻醇、单硬脂酸甘油酯、月桂基硫酸钠、二硬脂酸甘油酯单独或并用蜡,或本领域上公知的其他材料。
[0270]
根据要期望达成的化妆品性质,来选择适合用于配制剂的油类或脂肪,因为活性化合物在可能用于药物乳液配制剂中的大多数油类中的溶解度极低。因此,乳霜优选应为不油腻、不沾粘及可洗除的产品,具有合适的坚实度,以避免从管或其他容器中渗漏。可使用直链或分支链的单-或二元烷基酯如二-异己二酸酯、硬脂酸异鲸蜡基酯、椰子脂肪酸的丙二醇酯、肉豆蔻酸异丙基酯、油酸癸酯、棕榈酸异丙基酯、硬脂酸丁酯、棕榈酸2-乙基己基
酯或分支链酯的混合物。这些可以根据所需性质单独使用或联合使用。
[0271]
或者,可使用高熔点脂质,如白色软石蜡和/或液态石蜡或其他矿物油。
[0272]
适合表面施用至眼睛的配制剂亦包括滴眼液,其中活性成分已溶解或悬浮于合适载剂中,尤指活性成分的水性溶剂。
[0273]
活性成分于此类配制剂中的优选浓度为0.5至20%,有利地在0.5至10%,特别地约1.5%w/w。
[0274]
供胃肠外施用的配制剂可呈水性或非水性等渗无菌注射溶液或悬浮液形式。此等溶液及悬浮液可由无菌粉剂或粒剂,使用一种或多种上述用于调配经口施用的载剂或稀释剂制备,或使用其他合适匀散剂或湿化剂及悬浮剂制备。化合物可以溶于水、聚乙二醇、丙二醇、乙醇、玉米油、棉籽油、花生油、芝麻油、苯甲醇、氯化钠、黄芪胶和/或各种不同缓冲剂中。其他佐剂及施用模式是药物领域广泛已知者。活性成分亦可使用合适载剂(包括盐水、右旋糖、或水,或使用环糊精(亦即captisol)、共溶剂溶解法(亦即丙二醇)或胶束溶解法(亦即tween 80))形成组合物,供注射施用。
[0275]
无菌注射剂亦可为于无毒的胃肠外可接受的稀释剂或溶剂中的无菌注射溶液或悬浮液,例如,呈1,3-丁二醇的溶液。可使用的可接受的媒介物及溶剂为水、林格氏溶液及等渗性氯化钠溶液。此外,经常使用无菌的非挥发性油类作为溶剂或悬浮介质。基于此目的可以采用任何温和非挥发性油类,包括合成性单酸-或二酸甘油酯。此外,脂肪酸(如油酸)可用在注射制剂中。
[0276]
供肺部施用的药物组合物可呈气溶胶形式施用或使用包含干粉气溶胶的吸药器施用。
[0277]
供直肠施用药物的栓剂通过混合药物与合适的无刺激性赋形剂来制备,该赋形剂为诸如可可奶油及聚乙二醇,其在常温呈固体,但在直肠温度即呈液态,因此将会在直肠中熔化及释放药物。
[0278]
药物组合物可经过常规药物操作,如杀菌,且/或包含常用的佐剂,如防腐剂、安定剂、湿化剂、乳化剂、缓冲剂等等。片剂与丸剂可以另外使用肠溶性包衣制备。此类组合物亦可包含佐剂,如湿化剂、甜味剂、调味剂及香料剂。本发明药物组合物包含本文所述式的化合物,或其药学上可接受的盐;选自下列的额外药剂:激酶抑制剂(小分子、多肽、抗体等等)、免疫抑制剂、抗癌剂、抗病毒剂、消炎剂、抗真菌剂、抗生素、或抗血管过度增殖化合物;及任何药学上可接受的载剂、佐剂或媒介物。
[0279]
另一种本发明组合物包含本文所述式的化合物,或其药学上可接受的盐;及药学上可接受的载剂、佐剂或媒介物。此类组合物任选地包含一种或多种额外治疗剂,包括例如,激酶抑制剂(小分子、多肽、抗体等等)、免疫抑制剂、抗癌剂、抗病毒剂、消炎剂、抗真菌剂、抗生素、或抗血管过度增殖的化合物。
[0280]
术语“药学上可接受的载剂或佐剂”是指可本与本发明的化合物共同施用于患者的载剂或佐剂,及当以足够递送治疗量的化合物的剂量施用时,不会破坏其药理活性,且无毒。本发明药物组合物可使用的药学上可接受的载剂、佐剂及媒介物包括但不限于离子交换剂、矾土、硬脂酸铝、卵磷脂、自行乳化药物递送系统(self emulsifying drug delivery systems)(sedds)(如d-生育酚聚乙二醇1000琥珀酸盐)、用于药物剂型的界面活性剂(如tweens或其他类似的聚合递送基质)、血清蛋白质(如人血清白蛋白)、缓冲物质(如磷酸
盐)、甘氨酸、山梨酸、山梨酸钾、饱和植物性脂肪酸的部分甘油酯混合物、水、盐或电解质(如硫酸鱼精蛋白、磷酸氢二钠、磷酸氢钾、氯化钠、锌盐、胶体二氧化硅、三硅酸镁、聚乙烯吡咯烷酮、基于纤维素的物质、聚乙二醇、羧基甲基纤维素钠、聚丙烯酸酯、蜡类、聚乙二醇-聚氧丙烯-嵌段聚合物、聚乙二醇及羊毛脂。亦宜使用环糊精,如u-、p-及y-环糊精,或其化学修饰衍生物,如羟基烷基环糊精,包括2-及3-羟基丙基-环糊精,或其他可溶性衍生物,以增强递送本文所述式的化合物。
[0281]
药物组合物可呈任何口服可接受的剂型经口施用,包括但不限于胶囊、片剂、乳液及水性悬浮液、匀散液及溶液。以口服用片剂为例,常用的载剂包括乳糖及玉米淀粉。通常亦添加润滑剂,如硬脂酸镁。呈胶囊型式经口施用时,适用的稀释剂包括乳糖及干燥玉米淀粉。当经口施用水性悬浮液和/或乳液时,活性成分可以悬浮或溶解于油相中,与乳化剂和/或悬浮剂组合。
[0282]
若期望,可添加某些甜味剂、调味剂和/或着色剂。药物组合物可包括利用脂质体或微囊封技术的配制剂。其各种不同实例是本领域已知者。
[0283]
药物组合物可利用鼻气溶胶或吸入法施用。此类组合物是根据药物配制剂领域公知的技术制备,以及可于盐水中,使用苯甲醇或其他合适防腐剂、增强生物利用度的促进吸收剂,氟碳化物、和/或其他溶解剂或匀散剂制成溶液,其实例亦是本领域已知。
[0284]
9.治疗试剂盒
[0285]
本发明的一个方面涉及一种方便且有效进行根据本发明方法及用途的试剂盒。通常,药物包装物或试剂盒包含一个或多个容器,内部填充本发明药物组合物的一种或多种成分。此类试剂盒尤其适于递送口服用固体剂型,如片剂或胶囊。此类试剂盒优选包括许多单位剂量,且亦可包括卡片,让剂量以其预期使用顺序排列。若需要时,可提供帮助记忆的方式,例如,以数字、字母或其他标记的形式,或插入日历,标示治疗计划中可以施用剂量的日期。任选地与此类容器相关联的可以是由管理药品的制造、用法或贩售的政府机关规定的通知形式,该通知反映该机关已核准供人施用的制造、用法或贩售。
[0286]
下列代表性实施例包含重要的补充信息、范例及指南,其可适用于本发明的各种不同实施方案及其效物。这些实施例希望协助说明本发明,并且无意且不应构成限制本发明。的确,除了那些本文所示及描述者外,那些本领域技术人员在审查本案时,即可了解本发明各种不同修饰及其许多其他实施方案,包括下列实施例及本文所摘录科学与专利文献的参考文献。
[0287]
所摘录参考文献的内容已以引用的方式并入本文,以助于说明本领域。
[0288]
此外,针对本发明目的,化学元素是根据cas版的handbook of chemistryand physics,第75版内页的元素周期表鉴定。此外,一般有机化学原理及特定官能基部分体及反应性均描述于“organic chemistry”,thomas sorrell,university science books,sausalito:1999,及“organic chemistry,”morrison&boyd(第3版),此二者的完整内容已以引用的方式并入本文。
[0289]
10.合成方案
[0290]
本发明的化合物可由本领域普通技术人员根据本领域认可的技术及方法制备。更明确言之,本发明的化合物可以如下文所示反应方案、方法及实例制备。习此本领域者将了解,下列反应方案的个别步骤均可以变化,产生本发明的化合物。试剂及起始物很容易由习
此本领域者取得。除非另有明确说明,否则所有取代基均如前述定义。
实施例
[0291]
下列为说明书中所采用的缩写及其含义:
[0292]
ac:乙酰基
[0293]
boc:叔丁氧羰基
[0294]
etoac:乙酸乙酯
[0295]
dcm:二氯甲烷
[0296]
acn:乙腈
[0297]
thf:四氢呋喃
[0298]
dmso:二甲亚砜
[0299]
meoh:甲醇
[0300]
etoh:乙醇
[0301]
dmap:4-(二甲氨基)吡啶
[0302]
dipea:n,n-异丙基乙基胺
[0303]
nmr:核磁共振
[0304]
lc-ms:液相色谱-质谱
[0305]
tlc:薄层色谱法
[0306]
tcr:t细胞受体
[0307]
bcr:b细胞受体
[0308]
mm:毫摩尔
[0309]
μm:微摩尔
[0310]
ml:毫升
[0311]
ng:纳克
[0312]
nm:纳摩尔
[0313]
nm:纳米
[0314]
ic50:半数最大抑制浓度
[0315]
od:光密度
[0316]
a.生物实施例
[0317]
生物学实施例1 hpk1生化测定
[0318]
本实施例使用adp-glo
tm
激酶测定来测量潜在hpk1抑制剂化合物对hpk1激酶活性的影响。
[0319]
adp-glo
tm
激酶测定(promega corp.,madison,wi)测量激酶反应所形成的adp。根据制造商,激酶测定中形成的adp首先转化成atp,其接着用于在荧光素酶反应中产生光。发光度与激酶活性具相关性。下文描述了示例性实验设置,但个别测定中可能有些微调整。
[0320]
材料与仪器
[0321]
1.试剂:
[0322][0323]
2.仪器&供应商:
[0324][0325][0326]
3.板设置
[0327]
化合物从10μm(最高浓度)至0.508nm(最低浓度)的连续3倍稀释物。阳性对照为10μm参考物+酶+底物。阴性对照为1%dmso+酶+底物。
[0328]
4.规程
[0329]
1.缓冲液制备
[0330]
40mm tris ph7.5;20mm mgcl2,0.1mg/ml bsa,50μm dtt
[0331]
缓冲液储液
[0332]
1m tris,ph7.5,121.14g/mol添加6.057g至50ml h2o中,调整ph至7.5
[0333]
1m mgcl2,95.21g/mol添加4.7605g至50ml h2o中
[0334]
添加20ml 1m tris及10ml 1m mgcl2至470ml ddh2o中,得到缓冲液储液,并储存于室温。
[0335]
2.制备新鲜1*测定缓冲液
[0336]
试剂[储液][最终]倍数添加(ml)dtt(mm)100.052000.015
bsa(mg/ml)1000.110000.003缓冲液储液
ꢀꢀꢀ3[0337]
3.化合物制备
[0338]
1)通过将10μl 10mm各化合物储液与90μl dmso混合,稀释化合物至1mm。
[0339]
2)接着由bravo稀释化合物3倍成10种剂量(从5μl稀释至10μl)。最高化合物浓度为1mm(100
×
),dmso浓度为100%。
[0340]
3)通过echo将100nl各稀释化合物样品转移至384孔板(corning-4512)
[0341]
4)板于1,500rpm离心1分钟。
[0342]
4.制备2
×
atp-mbp混合物:
[0343]
20μm atp,0.2μg/μl mbp于激酶缓冲液(最终浓度:10μm atp,0.1μg/μl mbp)
[0344][0345]
5.使用测定缓冲液制备2
×
hpk1工作溶液。
[0346]
hpk1最终浓度为0.6ng/μl。高效力的化合物则使用较低浓度的hpk1(0.26ng/μl-0.065ng/μl)。
[0347]
6.添加5μl/孔的2
×
hpk1工作溶液,于1,500rpm离心1分钟。
[0348]
7.添加5μl/孔的2
×
atp-底物,于1,500rpm离心1分钟。
[0349]
8.于25℃温育1小时(或针对高效力化合物则为6小时)。
[0350]
9.添加5μl/孔adp-glo
tm
试剂中止激酶反应,并在1小时后消减未耗尽的atp。于1,500rpm离心1分钟。于25℃温育40min。
[0351]
10.添加10μl激酶检测试剂,使adp转化成atp。于1,500rpm离心1分钟。于25℃温育40min。
[0352]
11.于envision读板器(384-ctg)记录发光信号。
[0353]
5.数据分析
[0354]
根据及相对于包含在各测定板内的高对照与低对照中的信号,计算各化合物浓度的抑制百分比(%)。高对照孔作为0%抑制,及未包含任何化合物但仅包含dmso(最终浓度=0.5%)的低对照孔则作为100%抑制。以测试化合物的浓度及%抑制值绘图,并采用三参数逻辑剂量响应等式(three-parameter logistic dose response equation)确定50%抑制所需的化合物的浓度(ic50)。评估各实验中参考肽/化合物的终点值(ic50),作为质量控制测量值。若终点值在预期值的3倍之内,则该实验视为可接受的。
[0355]
生物学实施例2 pkc-theta生化测定
[0356]
本实施例采用adp-glo
tm
激酶测定来测量潜在hpk1抑制剂化合物对pkc-θ激酶活性的影响。下文描述了示例性实验设置,但个别测定中可能有些微调整。
[0357]
材料与仪器
[0358]
1.试剂:
[0359][0360]
2.仪器&供应商:
[0361][0362][0363]
3.板设置
[0364]
化合物从10μm(最高浓度)至0.508nm(最低浓度)的连续3倍稀释物。阳性对照为10μm参考物+酶+底物。阴性对照为1%dmso+酶+底物。
[0365]
4.规程
[0366]
1.缓冲液制备
[0367]
40mm tris ph7.5;20mm mgcl2,0.1mg/ml bsa,50μm dtt
[0368]
缓冲液储液
[0369]
1m tris,ph7.5,121.14g/mol添加6.057g至50ml h2o中,调整ph至7.5
[0370]
1m mgcl2,95.21g/mol添加4.7605g至50ml h2o
[0371]
添加20ml 1m tris及10ml 1m mgcl2至470ml ddh2o中,得到缓冲液储液,并储存于室温。
[0372]
2.制备新鲜1*测定缓冲液
[0373][0374]
3.化合物制备
[0375]
1)通过将10μl 10mm各化合物储液与90μl dmso混合,稀释化合物至1mm。
[0376]
2)接着由bravo稀释化合物3倍成10种剂量(从5μl稀释至10μl)。最高化合物浓度为1mm(100
×
),dmso浓度为100%。
[0377]
3)通过echo,将50nl各稀释化合物样品转移至384孔板(corning-4512)。
[0378]
4)板于1,500rpm离心1分钟。
[0379]
4.于激酶缓冲液中制备2
×
酶工作溶液:
[0380][0381]
5.制备2
×
atp-次混合物:
[0382][0383]
6.添加2.5μl/孔的2
×
酶工作溶液,于1,500rpm离心1分钟。
[0384]
7.添加2.5μl/孔的2
×
atp-底物,于1,500rpm离心1分钟。
[0385]
8.于25℃温育60min。
[0386]
9.添加5μl/孔的adp-glo
tm
试剂中止激酶反应,并在1小时后消减未耗尽的atp。于1,500rpm离心1分钟。于25℃温育40min。
[0387]
10.添加10μl激酶检测试剂,使adp转化成atp。于1,500rpm离心1分钟。于25℃温育40min。
[0388]
11.于envision读板器(384-usl)记录发光信号。
[0389]
5.数据分析
[0390]
根据及相对于包含在各测定板内的高对照与低对照中的信号,计算各化合物浓度
的抑制百分比(%)。高对照孔作为0%抑制,及未包含任何化合物但仅包含dmso(最终浓度=0.5%)的低对照孔则作为100%抑制。以测试化合物的浓度与%抑制值绘图,并采用三参数逻辑剂量响应等式确定50%抑制所需的化合物的浓度(ic50)。评估各实验中参考肽/化合物的终点值(ic50),作为质量控制测量值。若终点值在预期值的3倍之内,则该实验视为可接受的。
[0391]
生物学实施例3 tbk1生化测定
[0392]
本实施例采用adp-glo
tm
激酶测定来测量潜在hpk1抑制剂化合物对tbk1激酶活性的影响,下文描述了示例性实验设置,但个别测定中可能有些微调整。
[0393]
材料与仪器
[0394]
1.试剂:
[0395][0396]
2.仪器&供应商:
[0397]
仪器来源目录号96-v底孔板greiner651201384孔板corning4512ldv板labcytelp0200bravoagilent-envisionperkinelmer-水系统millipore milli-q参考系统-bravo 10μl尖端axygenvt-384-10ul-rbravo 30μl尖端axygenvt-384-31ul-r手动单通道移液管rainin-多通道电子移液管thermo/rainin-[0398]
3.板设置
[0399]
参见上文。
[0400]
4.规程
[0401]
1.缓冲液制备
[0402]
40mm tris ph7.5;20mm mgcl2,0.1mg/ml bsa,50μm dtt
[0403]
缓冲液储液
[0404]
1m tris,ph7.5,121.14g/mol
添加6.057g至50ml h2o中,调整ph至7.5
[0405]
1m mgcl2,95.21g/mol添加4.7605g至50ml h2o中
[0406]
添加20ml 1m tris及10ml 1m mgcl2至470ml ddh2o中,得到缓冲液储液,并储存于rt(室温)。
[0407]
2.制备新鲜1*测定缓冲液
[0408][0409]
3.化合物制备
[0410]
1)通过将10μl 10mm各化合物储液与90μl dmso混合,稀释化合物至1mm。
[0411]
2)接着由bravo稀释化合物3倍成10种剂量(从5μl稀释至10μl)。最高化合物浓度为1mm(100
×
),dmso浓度为100%。
[0412]
3)通过echo将50nl各稀释化合物样品转移至384孔板(corning-4512)。
[0413]
4)板于1,500rpm离心1分钟。
[0414]
4.于激酶缓冲液中制备2
×
酶工作溶液:
[0415][0416]
5.制备2
×
atp-次混合物:
[0417][0418]
6.添加2.5μl/孔的2
×
酶工作溶液,于1,500rpm离心1分钟。
[0419]
7.添加2.5μl/孔的2
×
atp-底物,于1,500rpm离心1分钟。
[0420]
8.于25℃温育60min。
[0421]
9.添加5μl/孔的adp-glo
tm
试剂中止激酶反应,并在1小时后消减未耗尽的atp。于1,500rpm离心1分钟。于25℃温育40min。
[0422]
10.添加10μl激酶检测试剂,使adp转化成atp。于1,500rpm离心1分钟。于25℃培养40min。
[0423]
11.于envision读板器(384-usl)记录发光信号。
[0424]
5.数据分析
[0425]
根据及相对于包含在各测定板内的高对照与低对照中的信号,计算各化合物浓度的抑制百分比(%)。高对照孔作为0%抑制,及未包含任何化合物但仅包含dmso(最终浓度=0.5%)的低对照孔则作为100%抑制。以测试化合物的浓度与%抑制值绘图,并采用三参数逻辑剂量响应等式确定50%抑制所需的化合物的浓度(ic50)。评估各实验中参考肽/化合物的终点值(ic50),作为质量控制测量值。若终点值在预期值的3倍之内,则该实验视为可接受的。
[0426]
生物学实施例4 jak3生化测定
[0427]
本实施例采用adp-glo
tm
激酶测定来测量潜在hpk1抑制剂化合物对jak3激酶活性的影响。下文描述了示例性实验设置,但个别测定法中可能有些微调整。
[0428]
材料与仪器
[0429]
1.试剂:
[0430][0431]
2.仪器&供应商:
[0432][0433][0434]
3.规程
[0435]
1.制备jak3激酶缓冲液
[0436]
新鲜添加dtt及bsa至缓冲液中(最终浓度:40mm tris ph7.5;20mm mgcl2,0.1mg/ml bsa,50μm dtt)。
[0437][0438]
2.化合物制备
[0439]
测试化合物:通过将5μl 10mm化合物储液与45μl dmso混合,稀释化合物至1mm。化合物溶液接着连续稀释3倍制成10种剂量。最高化合物浓度为1mm(100
×
),dmso浓度为100%。
[0440]
通过echo,将50nl的10种剂量的稀释化合物的每一种添加至384孔测定板(corning#4512)中。
[0441]
阳性对照:通过echo,将50nl 1mm参考化合物添加至384孔测定板(corning#4512)。
[0442]
阴性对照:将50nl dmso转移至384-孔测定板中,作为阴性对照。
[0443]
测定板于1,500rpm离心1分钟。
[0444]
3.根据下列程序,利用echo转移化合物:
[0445]
参考化合物与测试化合物1-15:从10μm至0.508nm的连续3倍稀释物(10种剂量);阴性对照为0.78ng jak3、4μm atp&0.2μg/μl poly、1%dmso;阳性对照为最高剂量的参考化合物、0.78ng jak3、0.2μg/μl poly(e4y1)、4μmatp、1%dmso。
[0446]
4.使用移液管(thermo,30μl多通道)添加2.5μl/孔的2
×
jak3工作溶液,于1,500rpm离心1分钟。
[0447]
5.使用移液管(thermo,30μl多通道)添加2.5μl/孔的2
×
atp-poly(e4y1)操作混合物,于1,500rpm离心1分钟。
[0448]
6.测定板于25℃温育60min。
[0449]
7.利用bravo添加5μl/孔的adp-glo
tm
试剂中止激酶反应,并在1小时后消减未耗尽的atp。于1,500rpm离心1分钟。于25℃温育40min。
[0450]
8.利用bravo添加10μl激酶检测试剂,使adp转化成atp。于1,500rpm离心1分钟。于25℃温育40min。
[0451]
9.于envision读板器(384-usl)记录发光信号。
[0452]
10.使用xl-fit处理数据。抑制%=[1-(测试孔-阴性对照)/(阳性对照-阴性对照)]*100%。
[0453]
5.数据分析
[0454]
根据及相对于包含在各测定板内的阴性对照与阳性对照中的信号,计算各化合物浓度的抑制百分比(%)。阴性对照孔作为0%抑制,及阳性对照孔则作为100%抑制。以测试化合物的浓度与%抑制值绘图,并采用三参数逻辑剂量响应等式确定50%抑制所需的化合
物的浓度(ic50)。评估各实验中参考肽/化合物的终点值(ic50),作为质量控制测量值。若终点值在预期值的3倍之内,则该实验视为可接受的。
[0455]
生物学实施例5 zap70生化测定
[0456]
本实施例采用adp-glo
tm
激酶测定来测量潜在hpk1抑制剂化合物对zap70激酶活性的影响。下文描述了示例性实验设置,但个别测定中可能有些微调整。
[0457]
材料及仪器
[0458]
1.试剂:
[0459][0460][0461]
2.仪器&供应商:
[0462][0463]
3.规程
[0464]
1.zap70激酶缓冲液制备:
[0465]
新鲜添加dtt及bsa至缓冲液中(最终浓度:40mm tris ph7.5;20mm mgcl2,0.1mg/
ml bsa,50μm dtt,2mm mncl2)。
[0466][0467][0468]
2.化合物制备:
[0469]
测试化合物:通过将5μl 10mm化合物储液与45μl dmso混合,将化合物首先稀释至1mm。这些化合物接着稀释3倍制成10种剂量。最高化合物浓度为1mm(100
×
),dmso浓度为100%。
[0470]
通过echo,将50nl化合物溶液转移至384孔测定板(corning#4512)中。
[0471]
阳性对照:通过echo,将50nl 1mm星孢菌素(staurosporine)转移至384孔测定板中,作为阳性对照。
[0472]
阴性对照:将50nl dmso转移至384孔测定板中,作为阴性对照。测定板于1,500rpm离心1分钟
[0473]
3.根据下列程序,利用echo转移化合物:
[0474]
星孢菌素与测试化合物1-15:从10μm至0.508nm的连续3倍稀释物(10种剂量);阴性对照为6.25ng zap70、10μm atp&0.4μg/μl poly、1%dmso;阳性对照为10μm星孢菌素、6.25ng zap70、10μm atp&0.4μg/μl poly、1%dmso。
[0475]
4.制备2
×
atp-poly(e4y1)混合物:20μm atp、0.8μg/μl poly(e4y1)于激酶缓冲液(最终浓度:10μm atp、0.4μg/μl poly(e4y1)。
[0476]
5.制备2
×
zap70工作溶液:(2.5ng/μl)于激酶缓冲液中(最终浓度为1.25ng/μl)。
[0477]
6.使用移液管(thermo,30μl多通道)添加2.5μl/孔的2
×
zap70工作溶液,于1,500rpm离心1分钟。
[0478]
7.使用移液管(thermo,30μl多通道)添加2.5μl/孔的2
×
atp-poly(e4y1)操作混合物,于1,500rpm离心1分钟。
[0479]
8.测定板于25℃温育60min。
[0480]
9.利用bravo添加5μl/孔的adp-glo
tm
试剂中止激酶反应,并在1小时后消减未耗尽的atp。于1,500rpm离心1分钟。于25℃温育40min。
[0481]
10.利用bravo添加10μl激酶检测试剂,使adp转化成atp。于1,500rpm离心1分钟。于25℃温育40min。
[0482]
11.于envision读板器(384-usl)记录发光信号。
[0483]
12.使用xl-fit处理数据。抑制%=[1-(测试孔-阴性对照)/(阳性对照-阴性对照)]*100%。
[0484]
5.数据分析
[0485]
根据及相对于包含在各测定板内的阴性对照与阳性对照中的信号,计算各化合物
浓度的抑制百分比(%)。阴性对照孔作为0%抑制,及阳性对照孔则作为100%抑制。以测试化合物的浓度与%抑制值绘图,并采用三参数逻辑剂量响应等式确定50%抑制所需的化合物的浓度(ic50)。评估各实验中参考肽/化合物的终点值(ic50),作为质量控制测量值。若终点值在预期值的3倍之内,则该实验视为可接受的。
[0486]
生物学实施例6 lck生化测定方案
[0487]
本实施例采用adp-glo
tm
激酶测定来测量潜在hpk1抑制剂化合物对lck激酶活性的影响。下文描述了示例性实验设定,但个别测定中可能有些微调整。
[0488]
材料及仪器
[0489]
1.试剂:
[0490][0491]
2.仪器&供应商:
[0492][0493]
[0494]
3.规程
[0495]
1.lck激酶缓冲液制备:
[0496]
新鲜添加dtt及bsa至缓冲液中(最终浓度:40mm tris ph7.5;20mm mgcl2,0.1mg/ml bsa,50μm dtt,2mm mncl2)。
[0497][0498]
2.化合物制备:
[0499]
测试化合物:通过将5μl 10mm化合物储液与45μl dmso混合,将化合物首先稀释至1mm。化合物溶液接着连续稀释3倍制成10种剂量。最高化合物浓度为1mm(100
×
),dmso浓度为100%。
[0500]
阳性对照:通过将3μl的10mm星孢菌素(staurosporine)储液与97μl dmso混合,稀释成300μm。
[0501]
采用bravo,将50nl化合物溶液及300μm星孢菌素转移至384-孔测定板(corning#4512)。
[0502]
阴性对照:将50nl dmso转移至384-孔测定板中,作为阴性对照。测定板于1,500rpm离心1分钟。
[0503]
3.根据以下程序,利用echo转移化合物:
[0504]
星孢菌素与测试化合物1-15:从10μm至0.508nm的连续3倍稀释物(10种剂量);阴性对照为7ng lck、20μm atp&0.4μg/μl poly、1%dmso;阳性对照为3μm星孢菌素、7ng lck、20μm atp&0.4μg/μl poly、1%dmso。
[0505]
4.制备2
×
atp-poly(e4y1)混合物:40μm atp、0.8μg/μl poly(e4y1)于激酶缓冲液(最终浓度:20μm atp、0.4μg/μl poly(e4y1))。
[0506]
5.制备2
×
lck工作溶液:(2.8ng/μl)于激酶缓冲液中(最终浓度为1.4ng/μl)。
[0507]
6.使用移液管(thermo,30μl多通道)添加2.5μl/孔的2
×
lck工作溶液,于1,000rpm离心1分钟。
[0508]
7.使用移液管(thermo,30μl多通道)添加2.5μl/孔的2
×
atp-poly(e4y1)操作混合物,于1,000rpm离心1分钟。
[0509]
8.测定板于25℃温育60min。
[0510]
9.利用bravo添加5μl/孔的adp-glo
tm
试剂中止激酶反应,并在1小时后消减未耗尽的atp。于1,000rpm离心1分钟。于25℃温育40min。
[0511]
10.利用bravo添加10μl激酶检测试剂,使adp转化成atp。于1,000rpm离心1分钟。于25℃温育30min。
[0512]
11.于envision读板器(384-usl)记录发光信号。
[0513]
12.采用xl-fit处理数据。抑制%=(阴性对照-试验孔)/(阴性对照-阳性对照)
×
100%。
[0514]
4.数据分析
[0515]
根据及相对于包含在各测定板内的阴性对照与阳性对照中的信号,计算各化合物浓度的抑制百分比(%)。阴性对照孔作为0%抑制,及阳性对照孔则作为100%抑制。以测试化合物的浓度与%抑制值绘图,并采用三参数逻辑剂量响应等式确定50%抑制所需的化合物的浓度(ic50)。评估各实验中参考肽/化合物的终点值(ic50),作为治疗控制测量值。若终点值在预期值的3倍至内,则该实验视为可接受的。
[0516]
生物学实施例7 map4k3蛋白激酶测定
[0517]
本实施例提供用于测量蛋白激酶map4k3对肽底物的磷酸化的测定方案。
[0518]
简言之,将map4k3、其底物和辅因子(atp及mg
2+
)于微量滴定板的孔中组合,并于25℃温育5小时。温育结束时,添加含edta的缓冲液中止反应。采用来自caliper life sciences的基于微流控的labchip 3000 drug discovery system进行底物和产物的分离和电泳法定量。
[0519]
针对此测定,map4k3底物为fam-gagrlgrdkyktlrqirq-nh2(fam为羧基荧光素)。肽底物通过毛细管电泳的纯度优选为》98%。
[0520]
典型测定设置及条件提供于下文。
[0521]
1.向384孔板的孔中添加5μl 2
×
酶缓冲液(或对照物)。
[0522]
2.添加100nl 100
×
化合物。若需要时,酶及化合物可在此时预温育。
[0523]
3.添加5μl 2
×
底物缓冲液。
[0524]
4.将板于25℃温育5小时。
[0525]
5.添加40μl 1.25
×
中止缓冲液中止反应。
[0526]
6.使用下表提供的数值,于caliper3000 drug discovery system作业。
[0527]
12个吸液芯片的分离条件
[0528]
初始延迟吸液时间50sec样品缓冲液后吸液时间40sec染料缓冲液后吸液时间40sec样品吸液时间0.2sec最终延迟吸液时间120sec染料吸液时间0.2sec压力-2psi下游电压-3000伏特上游电压-800伏特
[0529]
7.加载板,并开始进行电泳,使用蓝色激光(480nm)为激发光及使用绿色ccd(520nm)进行检测(ccd2)。
[0530]
以下列反应条件运转上述测定:共5小时;于25℃,在20mm 100%抑制剂edta存在下进行。
[0531]
最终测定反应混合物为:100mm hepes,ph 7.5;0.1%bsa;0.01%triton x-100;
1mm dtt;5mm mgcl2、10μm正钒酸钠、10μmβ-甘油磷酸酯、20μm atp;1%dmso(来自化合物);0.5μm fam-gagrlgrdkyktlrqirq-nh2;0.5nm map4k3酶。
[0532]
应注意,map4k3的比活性在批次间变化,并且酶浓度可能需要调整,使底物转化成产物的产率为约10-20%。
[0533]
使用labchip 3000毛细管电泳仪器进行电泳法,分离存在于各样品中的底物及产物肽。当底物及产物肽分离时,观察到两个荧光峰。以测定的底物与产物峰的相对荧光强度变化为参数,反映酶活性。采用hts well analyzer软件(caliper life sciences)分析毛细管电泳图(rda采集档案)。由总比率(psr):p/(s+p)确定各样品中的激酶活性,其中p为产物肽的峰高度,及s为底物肽的峰高度。
[0534]
各化合物在各种不同浓度(12种化合物浓度,间隔3
×
稀释度)测量酶活性。收集四次重复的阴性对照样品(0%-没有抑制剂下的抑制性)及阳性对照样品(100%-在20mm edta存在下的抑制性),并用于计算各化合物在各浓度下的%-抑制值。
[0535]
采用下列公式决定抑制百分比(p
inh
):
[0536]
p
inh
=(psr
0%-psr
inh
)/(psr
0%-psr
100
%)*100,其中psr
inh
为在抑制剂存在下的总比率,psr
0%
为没有抑制剂时的平均产物总比率,及psr
100%
为100%-抑制性对照样品的平均产物总比率。
[0537]
采用4参数s形剂量-响应模型,使用xlfit 4软件(ibds),拟合抑制曲线(p
inh
相对于抑制剂浓度),决定抑制剂的ic50值。
[0538]
该测定法中采用的某些材料与缓冲液列于下表中以供参考。
[0539]
[0540][0541]
生物学实施例8人pan t细胞所释放il2&ifn-γ的化合物功效研究
[0542]
本实施例为可以用于使用pan t细胞及elisa测定格式来确定化合物对il2&ifn-γ的释放影响的测定方法。
[0543]
材料及仪器
[0544]
1.试剂:
[0545]
[0546][0547]
2.仪器&供应商:
[0548][0549]
3.规程
[0550]
pan t分离及试剂制备的规程(第0天):
[0551]
1.细胞生长培养基:
[0552]
rpmi1640:atcc,目录号30-2001
[0553]
10%fbs:gibco,目录号10099141
[0554]
1%pen-strep:gibco,目录号15140122
[0555]
1%非必需氨基:gibco,目录号11140050
[0556]
β-巯基乙醇:gibco,目录号21985023
[0557]
2.pan t分离缓冲液制备:
[0558]
通过使用冲洗溶液将bsa储液溶液(#130-091-376)稀释
20倍,制备含有磷酸盐缓冲盐水(pbs)(ph 7.2)、0.5%牛血清白蛋白(bsa)及2mm edta的溶液。缓冲液在4℃下保存。缓冲液在使用前先脱气,因为气泡会阻滞柱。
[0559]
内容物[储液][最终浓度]倍数体积(ml)bsa10%0.5%205mledta0.5m2mm2500.4mlpbs,ph7.21
×1×
194.6ml总计
ꢀꢀꢀ
100ml
[0560]
3.冷冻pbmc的解冻
[0561]
1)培养基于37℃水浴中预热。
[0562]
2)细胞于37℃水浴中快速解冻。
[0563]
3)添加预温热的培养基至15ml试管中。将细胞转移至管中。
[0564]
4)以300
×
g离心8min.(离心提高0,下降0)。
[0565]
5)使用冲洗缓冲液洗涤pbmc。
[0566]
6)pbmc于300
×
g离心8min,洗涤2次(离心提高9,下降1)。
[0567]
7)细胞使用pan t细胞分离缓冲液再次悬浮,及计算细胞数。
[0568]
4.pan t细胞分离:
[0569]
使用微珠混合物(cocktail)染色细胞
[0570]
1)准备细胞及确定细胞数。使用70μm细胞过滤器过滤细胞。
[0571]
2)将细胞沉淀物再次悬浮,每107个总细胞数于40μl缓冲液中。
[0572]
3)每107个总细胞数添加10μl pant细胞生物素-抗体混合物。
[0573]
4)混合孔并于冰箱(冰)中温育10分钟。
[0574]
5)每107个总细胞数添加30μl缓冲液。
[0575]
6)每107个总细胞数添加20μl pan t细胞微珠混合物。
[0576]
7)混合孔并于冰箱(冰)中温育15分钟。
[0577]
8)进行后续磁性细胞分离法。
[0578]
注:a.动作快,保持细胞低温,及使用预冷溶液(2-8℃)。b.针对达成107总计细胞数的磁性标记体积。当使用较少细胞操作时,使用指示的相同体积。c.当使用较高细胞数操作时,随之放大所有试剂体积及总体积。d.为了最优性能,重点在于磁性标记之前先获得单一细胞悬浮液。
[0579]
随后的手动细胞分离法:
[0580]
1)将ls柱置入合适macs分离器的磁场中。其细节可参见各macs柱数据表。
[0581]
2)通过使用3ml缓冲液冲洗来准备柱。
[0582]
3)施加细胞悬浮液至柱。收集流过物,其代表已富集的t细胞。
[0583]
4)使用5ml缓冲液洗涤柱。收集已通过的未标记细胞,代表已富集的t细胞。
[0584]
提醒:一定要等到柱储槽已排空才继续进行下一个步骤。
[0585]
5.pan t细胞facs:
[0586]
1)分别取出50μl pbmc及pan t细胞至facs管中。
[0587]
2)细胞与抗人cd3/cd4/cd8抗体(1μl/1μl/1μl/管)于4℃温育20min。未染色的对照,将细胞与facs染色缓冲液温育。
[0588]
3)使用冷的染色缓冲液(含0.2%bsa及1mm edta的pbs)洗涤2次。
[0589]
4)运行facs。门控cd3
+
、cd3
+
cd4
+
和cd3
+
cd8
+
群,用于%分析。
[0590]
5)若pan t细胞的纯度高于90%时,则使用适当体积的细胞培养基稀释细胞悬浮液至1百万个细胞/ml。
[0591]
6)将细胞悬浮液分配至无菌可丢弃储槽中用于未来使用。
[0592]
用于制备化合物及抗人cd3/cd28的规程(第0天):
[0593]
1.化合物制备
[0594]
化合物连续稀释物(来源板1000
×
)
[0595]
使化合物溶解于100%dmso至浓度为10mm。然后3倍连续稀释成8个点的剂量。
[0596]4×
化合物剂量制备(中间板:corning-3599):
[0597]
于培养基中制备4
×
化合物溶液。上下吸取。对于zpe对照,在培养基中制备0.4%dmso(4
×
)。对于hpe对照,在培养基中制备0.4μm rgt003-026(4
×
)。
[0598]
2.抗人cd3(储液浓度6.76mg/ml):贮存在4℃。
[0599]
1)使用pbs稀释抗人cd3至最终浓度0.5μg/ml。
[0600]
2)除了阳性及阴性对照孔以外,在各孔中添加50μl/孔的cd3。阳性及阴性对照孔没有cd3/cd28刺激。
[0601]
3)于37℃的5%co2培养箱中温育2小时。
[0602]
4)从细胞培养板中除去50μl抗体溶液。每次使用200μl无菌pbs冲洗各孔2次。
[0603]
3.抗人cd28(4
×
)制备
[0604]
使用培养基稀释抗体,储液浓度为11.07mg/ml至2μg/ml(4
×
)。
[0605]
4.pma/离子霉素制备(4
×
)
[0606]
1)于培养基中稀释pma至400ng/ml(8
×
)。
[0607]
2)于培养基中稀释离子霉素至8μm(8
×
)。
[0608]
3)混合等体积的pma与离子霉素,得到4
×
混合物。
[0609]
用于刺激细胞的规程(第0天)
[0610]
1.将1
×
105个细胞/孔(100μl/孔)的细胞悬浮液转移至96孔板(细胞板:greiner-655180)。
[0611]
2.对于测试化合物及zpe/hpe对照,添加50μl/孔的抗人cd28。对于阳性或阴性对照,分别添加50μl/孔的4x pma/离子霉素溶液或培养基。
[0612]
3.根据下文出示的板作图,添加50μl/孔的化合物至细胞板中。对于zpe/hpe对照,分别添加50μl/孔的0.4%dmso溶液或0.4μm rgt003-026(4
×
)。对于阳性或阴性对照,添加50μl/孔的培养基。
[0613]
4.板温育48小时。
[0614]
用于il-2&ifn-γelisa的规程:
[0615]
第1天:包被板:
[0616]
1)每孔使用100μl于包被缓冲液中稀释的il-2&ifn-γ捕捉抗体包被微量孔。对于建议的抗体包被稀释物,参见批次特定说明书/分析证明书。密封板,并于4℃温育过夜。
[0617]
第2天:样品收集:
[0618]
1)于37℃与5%co2培养箱中温育48小时后,以1,000rpm离心细胞板10min。收集
100μl/孔上清液后,然后进行il-2及ifn-γelisa测定。上清液贮存在-80℃,并在次日进行il-2及ifn-γelisa测定。上清液可能需要稀释30至40倍,以确保测定不超过il-2及ifn-γ标准曲线的线性范围。
[0619]
2)添加新的培养基(含有抗cd28、p/i和化合物)至板中,100μl/孔。
[0620]
3)板作图:
[0621]
第3-4天:il-2&ifn-γelisa:
[0622]
1)吸出孔,并用≥300μl/孔的洗涤缓冲液洗涤3次。最后一次洗涤后,反转板并在吸水纸上吸干,以除去任何残留的缓冲液。
[0623]
2)使用≥200μl/孔测定稀释剂来封闭板。于室温温育1小时。
[0624]
3)如步骤2吸出/洗涤。
[0625]
4)于测定稀释剂中制备标准物。
[0626]
il-2标准储液制备:
[0627]
添加1ml去离子水至小瓶中(235ng/瓶),储液浓度为235ng/ml。以10μl/瓶等分标准储液,于-80℃冷冻。
[0628]
ifn-γ标准储液制备:
[0629]
添加1ml去离子水至小瓶中(145ng/瓶),储液浓度为145ng/ml。以10μl/瓶等分标准储液,于-80℃冷冻
[0630]
il-2/ifnγ的标准曲线的制备:
[0631]
将标准样品稀释至最高浓度为500pg/ml。然后运行2倍连续稀释成10个点剂量(包括空白对照)。将不同浓度的标准物转移至elisa板中,100μl/孔。
[0632]
1)吸取100μl的各标准物、样品和对照至适当孔中。密封板并于室温温育2小时。
[0633]
2)如步骤2吸出/洗涤,但共洗涤5次。
[0634]
3)添加100μl操作检测剂(检测抗体+sav-hrp试剂)至各孔中。密封板并于室温温育1小时。
[0635]
4)如步骤2吸出/洗涤,但共洗涤7次。注:在这个最后洗涤步骤中,每次洗涤均将孔浸泡在洗涤缓冲液中30秒至1分钟。
[0636]
5)添加100μl底物溶液至各孔中。板(没有板密封盖)于室温黑暗中温育30分钟。
[0637]
6)添加50μl中止反应溶液至各孔中。.
[0638]
7)在中止反应30分钟内,在450nm下读取吸光度。相对于570nm处的od值归一化od450nm。
[0639]
4.数据分析
[0640]
生化测定中,根据并相对于包含在各测定板内的hpe和zpe孔的信号,计算各化合物浓度的抑制百分比(%)。hpe孔视为100%有效,并且不包含任何化合物但包含dmso(最终浓度=0.1%)的zpe孔则视为0%有效。
[0641]
细胞测定法中,在各化合物浓度下计算il-2或ifn-γ相对于dmso对照的反应(未出示数据)。ec2x代表产生200%反应(2倍)时的化合物浓度,并采用graphpad prism计算ec50。
[0642]
生物学实施例9动力学溶解度测定
[0643]
本方案旨在测量测试样品在测定缓冲液中的动力学溶解度。该研究将按照国际生
物伦理标准“world medical association declaration of helsinki”进行。
[0644]
1.试剂
[0645]
对照:
[0646]
10mm普萘洛尔(propranolol),于dmso中
[0647]
10mm酮康唑(ketoconazole),于dmso中
[0648]
10mm三苯氧胺(tamoxifen),于dmso中
[0649]
去离子18.2m-cm mq超纯水(ultrapure water)
[0650]
磷酸,85%(h3po4)
[0651]
二甲亚砜
[0652]
测定缓冲液
[0653]
磷酸盐缓冲液,ph7.4
[0654]
2.规程
[0655]
校准曲线制备:
[0656]
300μm化合物溶液:将6μl化合物储备溶液添加到192μl的meoh/h2o(1:1)中。
[0657]
于meoh/h2o(1:1)中制备工作溶液
[0658][0659][0660]
1)以10mm浓度于dmso中制备测试化合物的储备溶液。在1.5ml eppendorf管中,将储备溶液一式三份稀释到100mm磷酸盐缓冲液中至100μm的目标浓度,最终dmso浓度为1%。
[0661]
2)将4μl的10mm dmso储备溶液添加至396μl的测定缓冲液中。
[0662]
3)在室温下保持样品管摇动(1000rpm)1小时。
[0663]
4)将样品管以12000rpm(eppendrof-5424,约13500g)离心10分钟,以沉淀未溶解的颗粒。
[0664]
5)将上清液以不同稀释度(未稀释,10倍稀释,100倍稀释,或其他倍数稀释)转移到新的管中。
[0665]
6)将5μl的工作溶液和稀释的上清液添加至95μl acn(含有is)。
[0666]
针对校准曲线通过lc-ms-ms分析样品。
[0667]
3.数据分析
[0668]
在甲醇/h2o(1:1)中制备标准校准曲线。温育1小时后由校准曲线确定上清液中的
测试化合物浓度,该校准曲线通过在0.02μm到60μm的动态范围内绘制峰面积与标称浓度来构建并且用于评估测试化合物的水溶性。
[0669]
生物学实施例10mdck渗透性测定
[0670]
此测定法仅用于生成筛选数据,并且预估候选药物的肠道吸收潜力。该研究将按照国际生物伦理标准“world medical association declaration of helsinki”进行。
[0671]
仪器
[0672]
◆
24-孔细胞培养板(pet膜):millipore
[0673]
◆
24-孔进料器托盘:millipore
[0674]
◆
移液器和管(eppendorf)
[0675]
◆
millicell ers系统
[0676]
◆
96-孔u形板(greiner)
[0677]
◆
96-孔微孔板(greiner)
[0678]
◆
37℃co2培养箱
[0679]
◆
flexstation 3(molecular devices)
[0680]
试剂
[0681]
细胞系:
[0682]
mdck(atcc)或mdck mdr1(nki)。
[0683]
细胞培养生长培养基(mem+10%fbs+1%neaa)。
[0684]
通过将50ml fbs和5ml neaa添加至445ml的mem中来制备生长培养基,或根据实际需要调整最终体积。
[0685]
胰蛋白酶-edta(invitrogen,cat#25200-072)。
[0686]
测定和给药溶液缓冲液。
[0687]
hanks平衡盐溶液(hanks balanced salt solution,hbss,invitrogen),含25mm hepes,ph7.4
[0688]
hanks平衡盐溶液(hbss,invitrogen)。
[0689]
[0690]
制备供体缓冲液:
[0691]
对于a到b方向:
[0692]
√含0.3%dmso和5μm萤光黄(lucifer yellow,ly)的hbss缓冲液:将150μl dmso和125μl ly(2mm)添加到50ml hbss缓冲液(ph 7.4)中。
[0693]
√含0.1%dmso和5μm萤光黄(ly)的hbss缓冲液:将50μl dmso和125μl ly(2mm)添加到50ml hbss缓冲液(ph 7.4)中。
[0694]
√含5μm萤光黄(ly)的hbss缓冲液:将125μl ly(2mm)添加到50ml hbss缓冲液(ph 7.4)中。
[0695]
对于b到a方向:
[0696]
√含0.3%dmso的hbss缓冲液:将150μl dmso添加到50ml hbss缓冲液(ph 7.4)中。
[0697]
√含0.1%dmso的hbss缓冲液:将50μl dmso添加到50ml hbss缓冲液(ph 7.4)中。
[0698]
√无dmso的hbss缓冲液。
[0699]
制备接收者缓冲液:
[0700]
对于a-到-b方向:制备含0.4%dmso的hbss缓冲液:将200μl dmso添加到50ml hbss缓冲液(ph7.4)中。
[0701]
对于b-到-a的方向:制备含0.4%dmso和5μm ly的hbss缓冲液:将200μl dmso和125μl ly(2mm)添加到50ml hbss缓冲液(ph 7.4)中。
[0702]
对照:
[0703]
使用以下公式制备10mm的于dmso中的化合物的储备浓度和于测定缓冲液中的萤光黄:
[0704]
(实际重量/分子量)/ml溶剂=10mm
[0705]
参考化合物:红霉素,美托洛尔,阿替洛尔
[0706][0707]
以4000rpm离心稀释的溶液5分钟。收集上清液用于化合物给药。
[0708]
制备用于标准曲线的化合物溶液(3μm/1μm/0.2μm/0.04μm/0.01μm/0.005μm):
[0709]
20
×
溶液:
[0710]
15μl(10mm)+485μl(meoh:h2o=1:1)—500μl(300μm)
[0711]
200μl(300μm)+800μl(meoh:h2o=1:1)
‑‑‑
1000μl(60μm)
[0712]
200μl(60μm)+400μl(meoh:h2o=1:1)
‑‑‑
600μl(20μm)
[0713]
200μl(20μm)+800μl(meoh:h2o=1:1)
‑‑‑
1000μl(4μm)
[0714]
200μl(4μm)+800μl(meoh:h2o=1:1)
‑‑‑
1000μl(0.8μm)
[0715]
200μl(0.8μm)+600μl(meoh:h2o=1:1)
‑‑‑
800μl(0.2μm)
[0716]
200μl(0.2μm)+200μl(meoh:h2o=1:1)
‑‑‑
400μl(0.1μm)
[0717]1×
溶液:3μl的20
×
溶液(0.1
–
60μm)+57μl 0.4%dmso hbss+60μl含is(200ng/ml osalmid)的acn
[0718]
研究设计
[0719]
◆
测试浓度:由发起人确定
[0720]
◆
温育温度和时间点:37℃,0和60分钟
[0721]
◆
样品规格:1-3
[0722]
规程
[0723]
常规培养和维持
[0724]
在mem+10%fbs+1%neaa中维持储用培养物,在75cm2组织培养物处理的烧瓶中生长并且每周分裂(传代)2次以维持期望的汇合。
[0725]
对于维持传代:将胰蛋白酶化细胞以1:20的标准传代比率分配到新烧瓶中。
[0726]
传代培养方案:
[0727]
√除出并丢弃培养基。
[0728]
√用0.25%(w/v)胰蛋白酶-0.53mm edta溶液冲洗细胞层两次,以除去所有含有胰蛋白酶抑制剂的血清痕迹。
[0729]
√向烧瓶中添加2.0至3.0ml胰蛋白酶-edta溶液并且在倒置显微镜下观察细胞,直到细胞层分散(通常在5至15分钟内)。
[0730]
(注意:为了避免结块,在等待细胞分离时不要通过敲击或摇晃烧瓶来搅动细胞。难以分离的细胞可以放置在37℃下以促进分散)。
[0731]
√添加6.0至8.0ml的完全生长培养基并且通过轻轻移液吸出细胞。
[0732]
√在新的培养容器中添加适当的等分的细胞悬浮液。
[0733]
√在37℃下温育培养物。
[0734]
接种测定板
[0735]
mdck测定板在运行测定前3-4天进行接种。24孔板以0.88
×
105/孔的细胞密度接种于400μl的顶部室体积(2.2
×
105/ml)中,以及向24孔基底室提供25ml体积的生长培养基。通常在测定前24小时对测定板提供生长培养基的更换。
[0736]
测定板的制备和跨上皮电阻(trans-epithelial electrical resistance,teer)测量
[0737]
mdck测定板在运行测定前用hbss+缓冲液进行冲洗。冲洗后,以400μl顶部室体积和0.8ml hbss+基底室体积将新鲜hbss+添加到测定板中。使用millicell ers系统欧姆表测量跨单层的电阻。(如果teer高于100欧姆*cm2,则将使用细胞)。
[0738]
标准曲线的制备
[0739]
细胞板的制备:
[0740]
◆
从顶部侧和基底侧除去缓冲液。根据板作图向顶部孔添加600μl的供体溶液(用
于a-到-b)或500μl的接收者溶液(用于b-到-a)。
[0741]
◆
通过向新的24孔板的孔添加800μl的接收者溶液(用于a-到-b)或900μl的供体溶液(b-到-a),制备新的基底板。
[0742]
◆
将顶部板和基底板放入37℃培养箱。
[0743]
分析板的制备:
[0744]
◆
5分钟后,将100μl样品从所有供体(对于a-到-b和b-到-a)转移到d0样品板的适当孔中。并将100μl样品从所有顶部室(a-到-b的供体和b-到-a的接收者)转移到萤光黄d0(d0ly)的微孔板的适当孔中。
[0745]
◆
将顶部板铺到基底板上,开始传送过程。
[0746]
◆
在90分钟,分离顶部板和基底板,并且将100μl样品从所有供体(对于a-到-b和b-到-a两者)转移到新的d90样品板的适当孔中,并将200μl样品从所有接收者转移到r90样品板的适当孔中。将100μl样品从所有基底室(a-到-b的接收者和b-到-a的供体)转移到新的萤光黄r90(r90ly)微孔板的适当孔中。
[0747]
◆
使用荧光读板器,通过在485nm的激发波长和535nm的发射波长下读取d0ly和r90ly来确定ly渗透性。
[0748]
◆
样品制备。
[0749]
◆
对于接收者溶液:60μl样品+60μl含is(200ng/ml osalmid)的acn。
[0750]
◆
对于供体溶液:6μl样品+54μl 0.4%dmso/hbss+60μl含is(200ng/ml osalmid)的acn。
[0751]
计算
[0752]
跨上皮电阻(teer)=(电阻
样品-电阻
空白
)
×
有效膜面积
[0753]
萤光黄渗透性
[0754]
p
app
=(va/(面积
×
时间))
×
([rfu]
接收者
–
[rfu]
空白
)/(([rfu]
初始,供体
–
[rfu]
空白
)
×
稀释因子)
×
100
[0755]
7.1使用以下等式进行药物运输测定制板:
[0756]
跨上皮电阻(teer)=(电阻
样品-电阻
空白
)
×
有效膜面积
[0757]
萤光黄渗透性
[0758]
p
app
=(vr/(面积
×
时间))
×
([rfu]
接收者
–
[rfu]
空白
)/(([rfu]
初始,供体
–
[rfu]
空白
)
×
稀释因子)
[0759]
药物渗透性
[0760]
p
app
=(vr/(面积
×
时间))
×
([药物]
接收者
/(([药物]
初始,供体
)
×
稀释因子)
[0761]
其中vr是接收者孔中的体积(a到b为0.8ml并且b到a为0.4ml),面积是膜的表面积(millipore-24细胞培养板为0.7cm2),并且时间是总传送时间,单位为秒。
[0762]
回收百分比=100
×
(在90分钟供体中的总化合物
×
稀释因子+在90分钟接收者中的总化合物)/(在0分钟供体中的总化合物
×
稀释因子)。
[0763]
数据分析
[0764]
使用以下等式计算测试化合物的表观渗透系数(p
app
):
[0765]
p
app
=(vr/(面积
×
时间))
×
([药物]
接收者
/(([药物]
初始,供体
)
×
稀释因子)
[0766]
其中vr是受体孔中的体积(a到b为0.8ml并且b到a为0.4ml),面积是膜的表面积
(millipore-24细胞培养板为0.7cm2),并且时间是总传送时间,单位为秒。接收者侧和供体侧的测试化合物浓度由标准校准曲线确定,该曲线是通过绘制峰面积与标称浓度构建。
[0767]
根据顶部到基底(a-b)p
app
数据和基底到顶部(b-a)p
app
数据的平均值计算出流出率(efflux ration):流出率=p
app(b-a)
/p
app(a-b)
。
[0768]
跨上皮电阻(teer)的测量用于确定细胞间的紧密连接的形成,并且如果teer值大于100ωcm2,则将使用细胞单层。为了评估细胞单层的完整性,将5μm的萤光黄与供体区室的测试化合物共处理。当共处理的萤光黄的p
app
大于5
×
10-6
cm/sec时,数据点将被排除在计算之外。
[0769]
生物学实施例11肝脏微粒体测定中的代谢稳定性
[0770]
此测定用于确定测试化合物在肝脏微粒体中的稳定性。该研究将按照国际生物伦理标准“world medical association declaration of”进行。
[0771]
肝脏微粒体样品
[0772]
人、sprague dawley大鼠、cd-1小鼠、beagle犬和cynomolgus猴肝脏微粒体样品购自bd gentest或rild。
[0773]
仪器
[0774]
培养箱(37℃)
[0775]
离心机(eppendorf 5810)
[0776]
eppendorf移液管和管
[0777]
96孔板(greiner)
[0778]
试剂
[0779]
化合物储备溶液:
[0780]
dmso中10mm测试化合物,在-80℃储存。
[0781]
测定缓冲液
[0782]
0.1m磷酸钾缓冲液,ph 7.4:
[0783]
缓冲液a:1.0l的0.1m含有1.0mm edta的磷酸二氢钾缓冲液
[0784]
缓冲液b:1.0l的0.1m含有1.0mm edta的磷酸氢二钾缓冲液
[0785]
缓冲液c(k-磷酸盐缓冲液):0.1m磷酸钾缓冲液,1.0mm edta,ph 7.4,通过用700ml缓冲液b与缓冲液a进行滴定同时监测ph计。
[0786]
加标溶液(spiking solution)
[0787]
制备3
×
测试化合物或阳性对照溶液:
[0788]
500μm加标溶液:将10μl的10mm dmso储备溶液添加到190μl acn中。
[0789]
微粒体(0.75mg/ml)中的1.5μm加标溶液:将1.5μl的500μm加标溶液和18.75μl的20mg/ml肝微粒体加入479.75μl的k-磷酸缓冲液中。
[0790]
其他溶液
[0791]
0.1m k-磷酸盐缓冲液中的6mm nadph。
[0792]
将25.1mg的nadph四钠盐溶解在5ml的k-磷酸盐缓冲液中。
[0793]
规程
[0794]
向指定45分钟、30分钟、15分钟、5分钟和0分钟的孔添加30μl含有0.75mg/ml微粒体溶液的1.5μm加标溶液。在37℃下预温育板5分钟。
[0795]
向所有的孔添加15μl nadph储备溶液(6mm),并开始计时。立即向指定为时间0的孔添加135μl的含有is的acn。
[0796]
在5分钟、15分钟、30分钟和45分钟,分别向孔添加135μl的含有is的acn。
[0797]
在温育结束时,向指定为时间0的孔添加15μl的nadph储备溶液(6mm)。
[0798]
淬灭后,将反应混合物3220
×
g离心10分钟。
[0799]
将每个孔中的50μl上清液转移到含有50μl超纯水(millipore)的96孔样品板中,用于lc/ms分析。
[0800]
表11-1最终温育混合物中组分的浓度
[0801][0802]
计算
[0803]
以每个时间点测试化合物的剩余%表示结果:
[0804]
剩余%=峰面积比t
x
/峰面积比t0,其中
[0805]
t0=0分钟的温育
[0806]
t
x
=任何给定的温育时间。
[0807]
体外半衰期按分钟报告并且按如下计算:
[0808]
t
1/2
=-(0.693/斜率)
[0809]
当在x=0分钟处y截距=100%时,斜率=ln(剩余%)vs温育时间。
[0810]
按如下根据t
1/2
计算体外内在清除率cl
′
int
。
[0811]
cl
′
int
=(0.693/t
1/2
)
×
(1/(微粒体蛋白浓度(0.5mg/ml))
×
比例因子
[0812]
表11-2中列出了用于每个物种的比例因子。
[0813]
每个物种的预测肝脏清除率是根据半衰期使用充分搅动模型计算。
[0814]
cl
hep
=(qh×
cl
′
int
×fub
)/(qh+cl
′
int
×fub
),其中
[0815]
qh是肝血流(ml/min/kg)(表11-2)。
[0816]fub
是血浆中未结合药物的片段,以及
[0817]
cl
′
int
是体外固有清除率。
[0818]
表11-2小鼠、大鼠、狗、猴和人微粒体中固有清除率预测的比例因子
[0819][0820]a比例因子=(每克肝脏的微粒体蛋白)
×
(每千克体重的肝脏重量)
[0821]
数据分析
[0822]
通过与t0(温育0分钟)处的峰面积比(化合物峰面积/内标峰面积)比较,计算出每个温育时间点的母体剩余%。峰面积比的ln值与时间作图并且确定线的梯度。根据相对峰面积vs时间的自然对数线性回归计算化合物的半衰期和代谢。使用以下公式计算微粒体中的半衰期和内在清除率(cl
int
)。
[0823]
体外t
1/2
(min)=0.693/清除速率常数(k)
[0824]
cl
int
(μl/min/mg的微粒体蛋白)=(0.693/体外t
1/2
)(温育体积/mg微粒体)。
[0825]
将使用充分搅动模型,根据计算的半衰期确定每个物种的测试化合物的预测清除率。
[0826]
测定中还将包含阳性对照。如果阳性对照不在规定限制内,将拒绝化合物的数值。
[0827]
生物学实施例12本公开化合物和类似物的比较
[0828]
根据本公开提供的方案,测试了本公开化合物和多种化合物类似物的生物学特性。测试结果汇总于以下表12-1和表12-2中。根据表1,本公开的化合物(实施例1、2、4和5)表现出优异的hpk1抑制活性(小于1nm)。它们在诱导il-2的产生方面也显示出良好的功效。此外,表12-2显示这些化合物在考虑溶解性、渗透性和代谢稳定性时具有良好的总体特性,其相比接近的类似物(化合物7-10)具有出乎意料总体更好的特性。
[0829]
表12-1 hpk1的ic
50
和功效
[0830]
[0831][0832]
表12-2动力学溶解度、渗透性和代谢稳定性
[0833]
[0834]
[0835][0836]
b.合成实施例
[0837]
仪器描述
[0838]
nmr光谱采用varian mercury光度计,于400mhz(1h)、376mhz(
19
f)或75mhz(
13
c)下操作。样品所采用的溶剂已于各化合物的实验方法中明确说明。化学位移以每百万分之一(ppm,δ单位)表示。偶联常数以赫兹单位(hz)表示。裂峰型态描述表观性多样性,并指定为s(单峰)、d(双峰)、t(三峰)、q(四峰)、quint(五峰)、m(多峰)、br(宽峰)。
[0839]
lcms采用下列系统:agilent 6120(二元梯度模组泵(binary gradient module pump)),xbridge分析柱c
18
,5μm,4.6
×
50mm,25℃,5μl注射体积,2ml/min,根据下列时程使用乙腈于0.1%乙酸铵水溶液中的梯度:
[0840]
时间(min)乙腈(%)0.1%乙酸铵水溶液(%)0.505954.509556.00955
[0841]
实验规程:所有反应均在无水氮气氛下进行,除非另有说明。tlc板采用紫外光目视检测。快速色谱法是指使用玻璃柱在硅胶上的柱色谱法(40-60μm)。或者,使用biotage sp1或biotage isolera系统进行自动化色谱法,于220或254nm进行u.v.检测,并采用biotage正相或逆相硅胶柱(silica cartridge)。其中细节可参见相关的实验方法。
[0842]
一般方法和制备
[0843]
本公开的化合物和中间体通过本文描述的合成法制备。实验过程中,除了明确述及的酸、碱、试剂、偶联试剂、溶剂等,对诸如温度、溶液浓度、溶剂体积、施加的微波条件、反应历时或其组合等反应条件的修饰均旨在成为本发明的一部分,可使用替代的合适酸、碱、试剂、偶联试剂、溶剂等,且均包括在本公开范围内。所有可能的几何异构体、立体异构体、盐形式均旨在落在本公开范围内。
[0844]
中间体
[0845]
int.0
[0846][0847]
3-甲基吲哚啉-1,3-二甲酸-1-(叔丁基)3-甲酯
[0848][0849]
步骤1:在25℃向dmf(8ml)中的吲哚啉-1,3-二甲酸-1-(叔丁基)3-甲酯(320mg,1.15mmol)和碘甲烷(491.36mg,3.46mmol)溶液添加氢化钠(50.77mg,1.27mmol,60%于矿物油中)。混合物在25℃搅动2小时,用ea(200ml)稀释,然后用水(20ml
×
3)和卤水(30ml)洗涤。有机层在无水na2so4上干燥并过滤。将滤液真空浓缩,并且通过硅胶上的快速柱色谱法(ea/pe:15%)纯化残余物以提供3-甲基吲哚啉-1,3-二甲酸-1-(叔丁基)3-甲酯,为浅黄色油(244mg,70%产率)。lc-ms(esi)m/z:236[m+h-56]
+
。
[0850]
int.1
[0851][0852]
2-(3-甲基吲哚啉-3-基)乙酸甲酯
[0853][0854]
步骤1:向thf(20ml)中的3-甲基吲哚啉-1,3-二甲酸-1-(叔丁基)3-甲酯(730mg,2.51mmol)溶液添加氢氧化钠(3.01g,75.17mmol)的水溶液(1.41ml)。混合物在室温下搅动16小时,用1m hcl水溶液酸化,并用etoac(100ml x 3)萃取。用卤水(50ml)洗涤有机相,在无水na2so4上干燥,并过滤。真空浓缩滤液以提供粗制1-叔丁氧羰基-3-甲基吲哚啉-3-羧酸,为黄色油(620mg,产率85%)。lc-ms(esi)m/z:222[m+h-56]
+
。
[0855]
步骤2:向dcm(15ml)中的1-叔丁氧羰基-3-甲基吲哚啉-3-羧酸(630mg,2.27mmol)的混合物缓慢加入0℃的草酰二氯(oxalyl dichloride,865.1mg,6.82mmol)和n,n-二甲基甲酰胺(8.3mg,0.114mmol)。混合物在室温下搅动16小时并且真空浓缩。将残余物(670mg,1.13mmol)在thf(10ml)和mecn(5.00ml)中溶解,并将重氮甲基(三甲基)硅烷(258.75mg,2.27mmol)(二乙醚中的2m溶液)缓慢加入上述溶液中。反应混合物在n2下搅动2小时,用10%柠檬酸(10ml)淬灭,并在dcm(50ml)和水(50ml)之间进行分区。有机相在无水硫酸钠上干燥并过滤。将滤液在真空中浓缩,并且通过硅胶上快速柱色谱法(ea/pe=0-10%)纯化残余物,以提供3-(2-重氮乙酰基)-3-甲基吲哚啉-1-甲酸叔丁酯,为黄色油(200mg,56%产率)。
[0856]
步骤3:向甲醇(5ml)中的3-(2-重氮乙酰基)-3-甲基吲哚啉-1-甲酸叔丁酯(200mg,0.664mmol)溶液添加苯甲酸银(silver benzoate,76.0mg,0.332mmol)。反应混合物在室温并且n2下搅动1.5小时,并用dcm(50ml)和水(50ml)稀释。分离有机层,在无水硫酸
钠上干燥,并过滤。滤液在真空中浓缩,并且通过硅胶上快速柱色谱法(ea/pe=0-15%)纯化残余物,以提供3-(2-甲氧基-2-氧代乙基)-3-甲基吲哚啉-1-甲酸叔丁酯,为无色油(100mg,42%产率)。lc-ms(esi)m/z:250[m+h-56]
+
。
[0857]
步骤4:将dcm(3ml)中的3-(2-甲氧基-2-氧代乙基)-3-甲基吲哚啉-1-甲酸叔丁酯(100mg,0.278mmol)和hcl/ea(4m,1.5ml)的混合物在室温下搅动2小时并真空浓缩,以提供粗制2-(3-甲基吲哚啉-3-基)乙酸甲酯,无需进一步提纯。lc-ms(esi)m/z:206[m+h]
+
。
[0858]
替代合成方法:
[0859][0860]
步骤1:在0℃下在30分钟内向thf(200ml)中的3-甲基吲哚啉-2-酮(20g,135.89mmol)溶液添加nah(6.52g,163.09mmol,60%悬浮于矿物油中)。将反应混合物在0℃搅动30分钟,然后滴加thf(50ml)中的碳酸二叔丁酯(29.07g,133.18mmol)溶液。将反应混合物搅动1小时,用饱和nh4cl水溶液(50ml)稀释,并用ch2cl2(100ml
×
2)萃取。合并的有机相在无水na2so4上干燥并过滤。滤液在真空中浓缩,并且通过硅胶上的快速柱色谱法(pe/ea=9/1)纯化残余物,以提供3-甲基-2-氧代吲哚啉-1-甲酸叔丁酯(30g,74%产率),为黄色油。lc-ms(esi)m/z:192[m+h]
+
。
[0861]
步骤2:在0℃下在30分钟内向thf(300ml)中的3-甲基-2-氧代吲哚啉-1-甲酸叔丁酯(30g,103.12mmol)溶液添加nah(4.95g,123.74mmol,60%于矿物油中)。反应混合物在0℃搅动30分钟,然后滴加2-溴乙酸甲酯(18.93g,123.74mmol)。反应混合物搅动1小时,用饱和nh4cl水溶液(100ml)淬灭,并用ch2cl2(200ml x 2)萃取。合并的有机相在无水na2so4上干燥并过滤。滤液在真空中浓缩,并且通过硅胶上的快速柱色谱法(pe/ea=8/2)纯化残余物,以提供3-(2-甲氧基-2-氧代乙基)-3-甲基-2-氧代吲哚啉-1-甲酸叔丁酯(28g,83%产率),为白色固体。lc-ms(esi)m/z:264[m+h-56]
+
。
[0862]
步骤3:将在1,4-二氧六环(35ml)中的4m hcl和ch2cl2(200ml)中的3-(2-甲氧基-2-氧代乙基)-3-甲基-2-氧代吲哚啉-1-甲酸叔丁酯(28g,87.68mmol)的混合物在室温下搅动2小时并真空浓缩。残余物在ch2cl2(200ml)和饱和nahco3水溶液(50ml)之间分区。分离的水层用ch2cl2(50ml
×
3)萃取,并且合并的有机层在无水na2so4上干燥并过滤。滤液在真空中浓缩,以提供2-(3-甲基-2-氧代吲哚啉-3-基)乙酸甲酯(17.6g,产率82%)。lc-ms(esi)m/z:220[m+h]
+
。
[0863]
步骤4:将在甲苯(220ml)中的2-(3-甲基-2-氧代吲哚啉-3-基)乙酸甲酯(15.6g,71.16mmol)和lawesson试剂(14.71g,36.38mmol)的混合物在130℃搅动1.5小时并真空浓缩。通过硅胶上的快速柱色谱法(pe/ea=80:20)纯化残余物,以提供2-(3-甲基-2-硫代吲哚啉-3-基)乙酸甲酯(15.5g,产率74%),为白色固体。lc-ms(esi)m/z:236[m+h]
+
。
[0864]
步骤5:在0℃下在1小时内向混合的thf(70ml)和甲醇(70ml)中的2-(3-甲基-2-硫
代吲哚啉-3-基)乙酸甲酯(9.5g,40.37mmol)和氯化镍(10.46g,80.75mmol)混合物中,分批加入nabh4(9.16g,242.24mmol)。将得到的混合物搅动10分钟并通过celite垫过滤。用meoh(100ml)洗涤固体饼,并真空浓缩滤液。在ea(200ml)中溶解残余物并用水(60ml
×
3)洗涤。有机相在无水硫酸钠上干燥并过滤。滤液在真空中浓缩,并且通过硅胶上的快速柱色谱法(ch2cl2:甲醇=95:5)纯化残余物,以提供2-(3-甲基吲哚啉-3-基)乙酸甲酯(6.5g,75%产率),为黄色油。lc-ms(esi)m/z:206[m+h]
+
。
[0865]
int.2
[0866][0867]
6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-胺
[0868][0869]
步骤1:在含2-(3-甲氧基苯基)乙烷胺(20.0g,132.27mmol,19.23ml)的hcl水溶液(1n,192ml,192mmol)混合物中添加甲醛水溶液(37%w.t.,41.64g,529.08mmol)。混合物于60℃搅动1小时,冷却至0℃,并在0℃滴加50%naoh水溶液(17.44g,218mmol)碱化。所得混合物于室温搅动过夜,并过滤。将滤液浓缩,产生双(6-甲氧基-3,4-二氢异喹啉-2(1h)-基)甲烷,为白色固体(23g,100%产率)。
[0870]
步骤2:于0℃,在含双(6-甲氧基-3,4-二氢异喹啉-2(1h)-基)甲烷(28g,82.73mmol)的i-proh(200ml)悬浮液中滴加浓hcl(15.20g,182.01mmol,15.3ml),混合物于室温搅动18小时。添加mtbe(70ml)至上述混合物中,悬浮液于室温再搅动4小时。过滤后,滤饼使用mtbe/i-proh混合物(100ml,1/1v/v)洗涤并干燥以提供6-甲氧基-1,2,3,4-四氢异喹啉盐酸盐的白色固体,其悬浮于dcm(300ml)中。在混合物中添加饱和nahco3水溶液(500ml),并且混合物于室温搅动2小时。分离后,水层使用dcm萃取(50ml x 4)。合并的有机层经无水na2so4干燥,过滤,并浓缩以提供6-甲氧基-1,2,3,4-四氢异喹啉,为黄色油(10g,75%产率)。lc-ms(esi)m/z:164[m+h]
+
。
[0871]
步骤3:于室温,在含6-甲氧基-1,2,3,4-四氢异喹啉(1.63g,9.99mmol)的meoh(30ml)溶液中添加甲醛水溶液(37%w/w,4.8g,59.92mmol,1.67ml)。混合物于室温搅动15分钟,并冷却至0℃,在其中分批添加nabh4(1.13g,29.96mmol)。混合物于室温搅动3小时,使用冰-水(10ml)中止反应,并使用dcm萃取(20ml x 5)。有机层经无水硫酸钠干燥,过滤,并浓缩。残余物经硅胶快速柱色谱法纯化(dcm/meoh=10/1v/v)以提供6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉,为黄色油(1.7g,96%产率)。lc-ms(esi)m/z:178[m+h]
+
。
[0872]
步骤4:于0℃,在浓硫酸的预冷却(0℃)溶液(4ml)中随后添加6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉(2g,11.28mmol)和硝酸胍(1.17g,9.59mmol)。所得混合物于0℃搅动30分钟,使用冰-水(20ml)中止反应,使用naoh水溶液(4n)碱化至ph 10-11,并使用dcm萃取(50ml x 4)。合并的有机层经无水na2so4干燥,过滤,并浓缩。残余物经硅胶快速柱色谱法纯化(pe 100%v/v,pe/ea=1/1v/v,ea100%v/v,并接着dcm/meoh=20/1,v/v)以提供6-甲氧基-2-甲基-7-硝基-1,2,3,4-四氢异喹啉,为黄色固体(0.9g,36%产率)。lc-ms(esi)m/z:
223[m+h]
+
。
[0873]
步骤5:在含6-甲氧基-2-甲基-7-硝基-1,2,3,4-四氢异喹啉(4.2g,14.40mmol)的ea(32ml)、h2o(16ml)和etoh(144ml)溶液中添加fe粉(5.5g,93.99mmol)和氯化铵(793.96mg,13.08mmol)。混合物于60℃搅动48小时,冷却至室温,并过滤。使用甲醇洗涤滤饼(30ml x 3),并将滤液浓缩。残余物经硅胶上的快速柱色谱法纯化(dcm/meoh=20/1至10/1v/v)以提供6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-胺盐酸盐的黄色固体(4.5g,100%产率)。lc-ms(esi)m/z:193[m+h]
+
。
[0874]
替代的合成方法:
[0875][0876]
步骤1:于0℃,在含三乙基胺(13.38g,132.27mmol,18.44ml)、2-(3-甲氧基苯基)乙烷胺(10g,66.13mmol)和dmap(807.96mg,6.61mmol)的dcm(100ml)溶液中慢慢添加boc2o(15.88g,72.75mmol,16.70ml)。混合物接着在室温搅动16小时,使用冰水(200ml)稀释并使用etoac萃取(3x200ml)。合并的有机相使用卤水(100ml)洗涤,经无水na2so4干燥,过滤,并真空浓缩。残余物经快速色谱法纯化(sio2,石油醚/乙酸乙酯=10/1)以提供(3-甲氧基苯乙基)氨基甲酸叔丁酯(14.6g,79.06%产率,90%纯度),为无色液体。lc-ms(esi)m/z:196[m+h-56]
+
。
[0877]
步骤2:于-78℃,在含(3-甲氧基苯乙基)氨基甲酸叔丁酯(7g,27.85mmol)和2-氯吡啶(4.74g,41.78mmol,3.92ml)的ch2cl2(50ml)溶液中添加tf2o(8.64g,30.64mmol,5.15ml)的ch2cl2(5ml)溶液。20分钟后,滴加bh3.et2o(19.77g,139.26mmol)至上述溶液中。反应混合物随后回升至室温,搅动2小时,并小心使用饱和nahco3溶液中止反应。所得混合物使用ch2cl2萃取(3x100ml)。合并的有机层使用卤水(50ml)洗涤,经无水na2so4干燥,过滤,并真空浓缩。残余物经快速色谱法纯化(sio2,石油醚/乙酸乙酯/甲醇=5/1/0至10/10/1)以提供6-甲氧基-3,4-二氢异喹啉-1(2h)-酮(3.4g,68.89%产率),为灰白色固体。lc-ms(esi)m/z:178[m+h]
+
。
[0878]
步骤3:于-20℃,在含6-甲氧基-3,4-二氢异喹啉-1(2h)-酮(3.8g,21.44mmol,9.62ml)的浓h2so4(50ml)溶液中滴加hno3(2.16g,22.29mmol,65%纯度)。混合物于-20℃~-25℃搅动3小时,倒至冰水(300ml)中。水层使用二氯甲烷萃取(3x200ml)。合并的有机层真空浓缩,并将残余物经快速柱色谱法纯化(sio2,石油醚/乙酸乙酯/甲醇=10/1/0~10/10/1)以提供6-甲氧基-7-硝基-3,4-二氢异喹啉-1(2h)-酮(2.02g,40.78%产率)。lc-ms(esi)m/z:223[m+h]
+
。
[0879]
步骤4:在含6-甲氧基-7-硝基-3,4-二氢异喹啉-1(2h)-酮(2.02g,9.09mmol)的thf(200ml)溶液中添加1m bh3/thf(45.46mmol,45.5ml)。混合物于回流下搅动20小时,并
小心使用甲醇(30ml)中止反应。所得溶液真空浓缩。残余物于2n hcl(50ml)中,于80℃加热3小时,冷却,使用氢氧化铵水溶液碱化,并使用二氯甲烷萃取(3x100ml)。合并的有机层经无水硫酸钠脱水,过滤,并浓缩以提供6-甲氧基-7-硝基-1,2,3,4-四氢异喹啉(1.89g,100.00%产率)。lc-ms(esi)m/z:209[m+h]
+
。
[0880]
步骤5:于室温,在含6-甲氧基-7-硝基-1,2,3,4-四氢异喹啉(1.89g,9.08mmol,9.62ml)的甲醇(30ml)溶液中添加甲醛(1.64g,54.46mmol,1.51ml)。混合物于室温搅动15分钟并冷却至0℃。然后分批添加nabh4(1.03g,27.23mmol)至上述混合物中。所得混合物于室温搅动3小时,使用冰水(10ml)中止反应,并使用dcm萃取(20ml x 5)。合并的有机层经无水硫酸钠脱水,过滤,并浓缩。残余物经硅胶快速柱色谱法纯化(dcm/meoh=10/1,v/v)以提供6-甲氧基-2-甲基-7-硝基-1,2,3,4-四氢异喹啉(1.4g,69.40%产率),为黄色油。lc-ms(esi)m/z:223[m+h]
+
。
[0881]
步骤6:在含6-甲氧基-2-甲基-7-硝基-1,2,3,4-四氢异喹啉(1.3g,5.85mmol)的etoac(2ml)、h2o(1ml)和etoh(10ml)溶液中添加fe(2.19g,39.19mmol)和氯化铵(284.74mg,5.32mmol)。混合物于60℃搅动16小时,冷却至室温,并过滤。使用甲醇洗涤饼(30ml x 3),并将滤液浓缩。残余物经硅胶快速柱色谱法纯化(dcm/meoh=20/1至10/1v/v)以提供6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-胺盐酸盐,为黄色固体(0.85g,75.6%产率)。lc-ms(esi)m/z:193[m+h]
+
。
[0882]
int.3
[0883][0884]
6-氟-2-甲基-1,2,3,4-四氢异喹啉-7-胺
[0885][0886]
步骤1:将h2so4(160ml)中的6-氟-3,4-二氢-2h-异喹啉-1-酮(20.0g,121.09mmol)的混合物冷却到-5℃,并分批加入硝酸钾(12.85g,127.15mmol)。将得到的混合物在同一温度下搅动4小时,并倒入冰水中。通过过滤收集固体,用水洗涤并真空干燥以得到6-氟-7-硝基-3,4-二氢-2h-异喹啉-1-酮(25.2g,119.91mmol,99.0%产率),为白色固体。1h nmr(400mhz,dmso-d6)δ8.46(d,j=7.9hz,1h),8.33(d,j=27.5hz,1h),7.63(d,j=11.8hz,1h),3.43(td,j=6.6,2.8hz,2h),3.04(t,j=6.5hz,2h).lcms(esi
+
)[(m+h)
+
]:211。
[0887]
步骤2:向6-氟-7-硝基-3,4-二氢-2h-异喹啉-1-酮(25.2g,119.91mmol)添加1m硼烷四氢呋喃(599.54mmol,600ml)。混合物在回流下搅动20小时,冷却到室温并用甲醇(150ml)小心地淬灭。所得溶液在真空中浓缩。在80℃将残余物在2n hcl水溶液(500ml)中搅动3小时,冷却至室温,用氢氧化铵溶液碱化至ph8~9,并用二氯甲烷(500ml
×
3)萃取。合并的有机层在无水硫酸钠上干燥并过滤。真空浓缩滤液以提供6-氟-7-硝基-1,2,3,4-四氢异喹啉(22.3g,113.67mmol,94.8%产率),为黄色固体。lcms(esi
+
)[(m+h)
+
]:197。
[0888]
步骤3:在室温下向dcm(500ml)中的6-氟-7-硝基-1,2,3,4-四氢异喹啉(22.3g,113.67mmol)溶液添加甲醛(formaldehyde,20.48g,682.03mmol)。混合物在室温下搅动30分钟并冷却至0℃。向该混合物分次添加三乙酰氧基硼氢化钠(96.37克,454.69mmol)。所得混合物在室温下搅动36小时,用冰水(400ml)淬灭并用dcm(600ml
×
3)萃取。合并的有机层在无水硫酸钠上干燥并过滤。滤液在真空中浓缩,并且通过硅胶上的快速柱色谱法(dcm/meoh=10/1v/v)纯化残余物,以提供6-氟-2-甲基-7-硝基-3,4-二氢-1h-异喹啉(21.3g,101.33mmol,89.1%产率),为黄色油。lcms(esi
+
)[(m+h)
+
]:211。
[0889]
步骤4:向etoh(213ml)和h2o(40ml)中的6-氟-2-甲基-7-硝基-3,4-二氢-1h-异喹啉(21.3g,101.33mmol)溶液添加氯化铵(37.22g,709.31mmol)和铁粉(56.59克,1.01mol,7.20ml)。混合物在60℃搅动3小时,冷却至室温并过滤。用甲醇(100ml
×
2)洗涤固体滤饼,并且真空浓缩滤液。通过硅胶上快速柱色谱法(dcm/meoh=20/1至10/1v/v)纯化残余物,以提供6-氟-2-甲基-3,4-二氢-1h-异喹啉-7-胺(17.3g,95.99mmol,94.7%产率),为黄色固体。lcms(esi
+
)[(m+h)
+
]:181.
[0890]
实施例1
[0891][0892]
2-(1-(5-氯-2-((6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸
[0893]
步骤1:向n-buoh(3ml)中的int.1(67mg,0.248mmol)的混合物缓慢添加2,4,5-三氯嘧啶(45.5mg,0.248mmol)和dipea(96.19mg,0.744mmol)。将混合物在100℃下搅动16小时,用冰水(20ml)淬灭并用ea(30ml
×
3)萃取。用卤水(20ml)洗涤有机相,在无水na2so4上干燥,并过滤。滤液在真空中浓缩,并且通过硅胶上快速柱色谱法(dcm/pe=0-100%)纯化残余物以得到2-(1-(2,5-二氯嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸甲酯(80mg,83.3%产率),为黄色固体。lc-ms(esi)m/z:352[m+h]
+
。
[0894]
步骤2:向i-proh(5ml)中的2-(1-(2,5-二氯嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸甲酯(80mg,0.227mmol)混合物缓慢添加int.2(51.5mg,0.227mmol)和tsoh h2o(43.2mg,0.227mmol)。将混合物在100℃下搅动16小时,用饱和nahco3水溶液(10ml)淬灭并用dcm/meoh(v/v=10/1,30ml
×
3)萃取。用卤水(30ml)洗涤有机相,在无水na2so4上干燥,并过滤。滤液在真空中浓缩,并且通过硅胶上快速柱色谱法(dcm/meoh=0-15%)纯化残余物,以得到2-(1-(5-氯-2-((6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)3-甲基吲哚啉-3-基)乙酸甲酯(70mg,42.5%yield)as yellow solid.lc-ms(esi)m/z:508[m+h]
+
.1h nmr(500mhz,dmso-d6)δ8.26(s,1h),7.98(s,1h),7.59(s,1h),7.30(t,j=8.8hz,2h),7.12(s,1h),6.98(s,1h),6.74(s,1h),4.38(d,j=10.7hz,1h),4.05(d,j=10.7hz,1h),3.78(s,3h),3.50(s,3h),3.27(s,2h),2.78(dd,j=10.3,4.8hz,3h),2.68(s,1h),2.55(t,j=5.7hz,2h),2.30(s,3h),1.36(s,3h)。
[0895]
步骤3:将thf(5ml)中2-(1-(5-氯-2-((6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸甲酯(70mg,0.096mmol)和lioh水溶液
(2n,1ml)的混合物在室温下搅动16小时,用甲酸调节至ph~5,并用dcm(30ml x 3)萃取。合并的有机相在无水硫酸钠上干燥并过滤。真空浓缩滤液,并且通过制备型hplc(mecn/10mm nh4hco3,0.025%nh3·
h2o)纯化残余物,以得到2-(1-(5-氯-2-((6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸(23.0mg,46.2%产率),为白色固体。lc-ms(esi)m/z:494[m+h]
+
.1h nmr(500mhz,dmso-d6)δ8.25(s,1h),7.99(s,1h),7.57(s,1h),7.29(dd,j=17.2,7.6hz,2h),7.11(t,j=7.3hz,1h),6.98(d,j=7.3hz,1h),6.75(s,1h),4.42(d,j=10.7hz,1h),4.02(d,j=10.6hz,1h),3.78(s,3h),3.30(s,2h),2.78(s,2h),2.69(d,j=15.4hz,1h),2.58(d,j=6.1hz,3h),2.32(s,3h),1.34(s,3h)。
[0896]
实施例2和3
[0897][0898]
(r)-2-(1-(5-氯-2-((6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸和(s)-2-(1-(5-氯-2-((6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸
[0899]
方法1:
[0900]
步骤1:sfc手性分离2-(1-(5-氯-2-((6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸甲酯(157mg),以分别提供(r)-2-(1-(5-氯-2-((6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸甲酯和(s)-2-(1-(5-氯-2-((6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸甲酯(67mg、63mg)。立体化学不是绝对确定的。
[0901]
对应峰1的组分:(r)-2-(1-(5-氯-2-((6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸甲酯或(s)-2-(1-(5-氯-2-((6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸甲酯。
[0902]
lc-ms(esi)m/z:508[m+h]
+
。手性-hplc保留时间:1.829分钟;ee值:98.3%。
[0903]
对应峰2的组分:(s)-2-(1-(5-氯-2-((6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸甲酯或(r)-2-(1-(5-氯-2-((6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸甲酯。
[0904]
lc-ms(esi)m/z:508[m+h]
+
。手性-hplc保留时间:3.427min;ee值:》99%。
[0905]
sfc分离条件:
[0906]
仪器:sfc-80(thar,waters)
[0907]
柱:ad-h 20*250mm,10μm(daicel)
[0908]
柱温:35℃
[0909]
流动相:co2/etoh(0.2%甲醇/氨)=50/50
[0910]
流动速率:80g/min
[0911]
背压:100bar
[0912]
检测波长:214nm
[0913]
循环时间:6.9min
[0914]
步骤2:在与实施例1的步骤3相同的条件下分别水解(r)-2-(1-(5-氯-2-((6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸甲酯和(s)-2-(1-(5-氯-2-((6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸甲酯,以提供(r)-2-(1-(5-氯-2-((6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸和(s)-2-(1-(5-氯-2-((6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸(分别为17.6mg,33.8mg)。
[0915]
实施例2:((r)-2-(1-(5-氯-2-((6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸甲酯或(s)-2-(1-(5-氯-2-((6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸甲酯对应于衍生自方法1的步骤1中sfc分离的峰1的产物。
[0916]
lc-ms(esi)m/z:494[m+h]
+
.1h nmr(400mhz,dmso-d6)δ8.25(s,1h),8.00(s,1h),7.57(s,1h),7.30(dd,j=13.7,7.7hz,2h),7.11(t,j=7.6hz,1h),6.97(t,j=7.3hz,1h),6.75(s,1h),4.42(d,j=10.7hz,1h),4.02(d,j=10.8hz,1h),3.78(s,3h),3.30(s,2h),2.78(s,2h),2.69(d,j=15.4hz,1h),2.62-2.54(m,3h),2.32(s,3h),1.34(s,3h).手性-hplc保留时间:1.384min;ee值:》99%。
[0917]
实施例3:((s)-2-(1-(5-氯-2-((6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸甲酯或(r)-2-(1-(5-氯-2-((6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸甲酯对应于衍生自方法1的步骤1中sfc分离的峰2的产物。
[0918]
lc-ms(esi)m/z:494[m+h]
+
.1h nmr(400mhz,dmso-d6)δ8.25(s,1h),8.00(s,1h),7.56(s,1h),7.29(dd,j=13.6,7.4hz,2h),7.10(t,j=7.1hz,1h),6.97(t,j=7.4hz,1h),6.75(s,1h),4.41(d,j=10.8hz,1h),4.02(d,j=10.8hz,1h),3.77(s,3h),3.29(s,2h),2.77(t,j=5.6hz,2h),2.69(d,j=15.4hz,1h),2.57(dd,j=13.1,7.1hz,3h),2.30(d,j=11.1hz,3h),1.34(s,2h).手性-hplc保留时间:1.132min;ee值:》99%。
[0919]
方法2:
[0920]
步骤1:sfc手性分离2-(1-(2,5-二氯嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸甲酯,以提供(r)-2-(1-(2,5-二氯嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸甲酯和(s)-2-(1-(2,5-二氯嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸甲酯。
[0921]
对应峰1的组分:lc-ms(esi)m/z:352[m+h]
+
。手性-hplc保留时间:1.191min(峰1);ee值:》99%。
[0922]
对应峰2的组分:lc-ms(esi)m/z:352[m+h]
+
。手性-hplc保留时间:1.519min(峰2);ee值:》99%。
[0923]
sfc分离条件:
[0924]
仪器:sfc-80(thar,waters)
[0925]
柱:ad-h 20*250mm,10μm(daicel)
[0926]
柱温:35℃
[0927]
流动相:co2/异丙醇(0.2%甲醇/氨)=87/13
[0928]
流动速率:80g/min
[0929]
背压:100bar
[0930]
检测波长:214nm
[0931]
循环时间:2.8min
[0932]
在类似或相同的条件下,对2-(1-(2,5-二氯嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸甲酯进行额外大批量的sfc手性分离,以提供(r)-2-(1-(2,5-二氯嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸甲酯和(s)-2-(1-(2,5-二氯嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸甲酯。
[0933]
(作为步骤2&3的代表实例的一个立体异构体)
[0934]
步骤2:将叔丁醇(25ml)中的int.2(1.36g,5.96mmol)、(s)-2-(1-(2,5-二氯嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸甲酯或(r)-2-(1-(2,5-二氯嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸甲酯(2.1g,5.96mmol,其衍生自方法2的步骤1的sfc分离的峰2),和tsoh h2o(1.13g,5.96mmol)的混合物在100℃下搅动12小时,冷却至室温,倒入饱和nahco3水溶液(10ml)中并用ea(30ml
×
3)萃取。有机相在无水na2so4上干燥并过滤。滤液在真空中浓缩,并且通过硅胶上快速柱色谱法(dcm/meoh=20:1)纯化残余物,以提供(s)-2-(1-(5-氯-2-((6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸甲酯或(r)-2-(1-(5-氯-2-((6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸甲酯(1.8g,产率56.3%),为浅黄色固体。lc-ms(esi)m/z:508[m+h]
+
。
[0935]
步骤3:将水中的氢氧化锂水合物(264mg,6.3mmol)的溶液和meoh(5ml)中的(s)-2-(1-(5-氯-2-((6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸甲酯或(r)-2-(1-(5-氯-2-((6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸甲酯(1.6g,3.15mmol)的混合物在室温下搅动16小时,并用乙酸酸化至ph约3。通过制备型hplc(mecn/10mm nh4hco3)纯化所得混合物,以提供实施例3:((s)-2-(1-(5-氯-2-((6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸或(r)-2-(1-(5-氯-2-((6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸)(839.3mg,产率53.9%),为浅黄色固体,其衍生自方法2的步骤1的sfc分离的峰2。lc-ms(esi)m/z:494[m+h]
+
.1h nmr(400mhz,dmso-d6)δ8.30(s,1h),8.02(s,1h),7.63(s,1h),7.37(d,j=8.0hz,1h),7.33(d,j=8.0hz,1h),7.16(t,j=8.0hz,1h),7.02(t,j=8.0hz,1h),6.80(s,1h),4.48(d,j=10.8hz,1h),4.08(d,j=10.8hz,1h),3.83(s,3h),2.83(t,j=5.6hz,2h),2.74(d,j=15.2hz,1h),2.66
–
2.59(m,3h),2.37(s,3h),1.40(s,3h).手性-hplc保留时间:1.132min;ee值:》99%。
[0936]
实施例4
[0937]
[0938]
2-(1-(5-氯-2-((6-氟-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸
[0939]
步骤1:将含2-(1-(2,5-二氯嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸甲酯(195.4mg,0.555mmol)、int.3(100mg,0.555mmol)和tsoh.h2o(105.5mg,0.555mmol)的i-proh(4ml)溶液于120℃搅动16小时,冷却至室温,并使用饱和nahco3水溶液中和至ph约8。混合物使用dcm/meoh萃取(v/v,10/1,20ml x3)。有机相经无水硫酸钠干燥,并过滤。将滤液真空浓缩,并且残余物经硅胶快速柱色谱法纯化(dcm/meoh=10/1),获得2-(1-(5-氯-2-((6-氟-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸甲酯与2-(1-(5-氯-2-((6-氟-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸异丙酯的混合物(440mg),为淡黄色固体,其用于下一个步骤而无需进一步纯化。lc-ms(esi)m/z:496(甲酯),524(异丙酯)[m+h]
+
。
[0940]
步骤3:将在thf(4ml)中的2-(1-(5-氯-2-((6-氟-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸甲酯与2-(1-(5-氯-2-((6-氟-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸异丙酯的混合物(220mg)以及lioh水溶液(2n,4ml)的混合物于室温搅动5小时并于50℃搅动16小时,使用甲酸调整至ph约5,并使用dcm/meoh萃取(10/1,20ml x 3)。合并的有机相经无水硫酸钠脱水,并过滤。将滤液真空浓缩,并且残余物经制备型hplc纯化(mecn/10mm nh4hco3),获得2-(1-(5-氯-2-((6-氟-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)-3-甲基吲哚啉-3-基)乙酸(26.5mg,12.4%产率)的白色固体。lc-ms(esi)m/z:482[m+h]
+
。1h nmr(400mhz,dmso-d6)δ8.93(s,1h),8.22(s,1h),7.35-7.24(m,3h),7.07-6.92(m,3h),4.41(d,j=10.7hz,1h),4.03(d,j=10.8hz,1h),3.36(s,2h),2.78(d,j=5.4hz,2h),2.68(d,j=15.5hz,1h),2.60-2.54(m,3h),2.32(s,3h),1.33(s,3h)。
[0941]
实施例5和6
[0942][0943]
遵循用于实施例2和3的方法2。
[0944]
步骤1:向在正丁醇(无水,99.9%,600ml)中的实施例2和3的方法2中所述的(峰1)((r)-2-[1-(2,5-二氯嘧啶-4-基)-3-甲基-吲哚啉-3-基]乙酸甲酯或(s)-2-[1-(2,5-二氯嘧啶-4-基)-3-甲基-吲哚啉-3-基]乙酸甲酯)(27.8g,78.93mmol)的第一洗脱物添加6-氟-2-甲基-3,4-二氢-1h-异喹啉-7-胺(int.3,17.07g,94.71mmol)和4-甲基苯磺酸(14.95g,86.82mmol)。混合物在120℃下搅动20小时。通过lcms判断反应完成后,将反应混合物冷却至室温,用饱和nahco3水溶液(100ml)淬灭,并用dcm(600ml
×
3)萃取。有机相在无水na2so4上干燥并过滤。滤液在真空中浓缩,通过硅胶上的快速柱色谱法(dcm/meoh=50/1至10/1v/v)纯化残余物,以提供(r)-2-[1-[5-氯-2-[(6-氟-2-甲基-3,4-二氢-1h-异喹啉-7-基)氨基]嘧啶-4-基]-3-甲基-吲哚啉-3-基]乙酸甲酯或(s)-2-[1-[5-氯-2-[(6-氟-2-甲基-3,
4-二氢-1h-异喹啉-7-基)氨基]嘧啶-4-基]-3-甲基-吲哚啉-3-基]乙酸甲酯(22.6g,45.57mmol,产率57.7%,与丁酯混合),为黄色固体。lcms(esi
+
)[(m+h)
+
]:496。
[0945]
步骤2:将在thf(200ml)和meoh(100ml)中的(r)-2-[1-[5-氯-2-[(6-氟-2-甲基-3,4-二氢-1h-异喹啉-7-基)氨基]嘧啶-4-基]-3-甲基-吲哚啉-3-基]乙酸甲酯或(s)-2-[1-[5-氯-2-[(6-氟-2-甲基-3,4-二氢-1h-异喹啉-7-基)氨基]嘧啶-4-基]-3-甲基-吲哚啉-3-基]乙酸甲酯(22.6g,45.57mmol)和氢氧化锂水溶液(5.46g,227.83mmol)(1n在h2o中)的混合物在25℃搅动6小时,直至lcms指示反应完全。在0℃用fa调节ph至5~6,并用dcm中的10%meoh(500ml
×
3)萃取。有机相在无水na2so4上干燥并过滤。真空浓缩滤液,并且通过制备型hplc(nh4hco3)纯化残余物(20g,80-90%纯度),以得到实施例5((r)-2-[1-[5-氯-2-[(6-氟-2-甲基-3,4-二氢-1h-异喹啉-7-基)氨基]嘧啶-4-基]-3-甲基-吲哚啉-3-基]乙酸或(s)-2-[1-[5-氯-2-[(6-氟-2-甲基-3,4-二氢-1h-异喹啉-7-基)氨基]嘧啶-4-基]-3-甲基-吲哚啉-3-基]乙酸)(9.6g,19.92mmol,43.7%产率),为浅黄色固体。lcms(esi
+
)[(m+h)
+
]:482。旋光度:-7.24(溶剂:can中的1%dea:水=50:50;浓度:0.3g/100ml;温度:25℃)。手性-hplc保留时间:13.962min(柱条件:columnig(4.6
×
250mm 5μm);流动相:正己烷(0.1%dea):etoh(0.1%dea)=80:20;波长:225nm;流速:1.0ml/min;温度:40℃)。实施例5对应于从实施例2和3的方法2中步骤1的sfc分离的峰1衍生的产物,其导致更有力的产物。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1