作为HPK1调节剂的嘧啶及吡啶衍生物和其使用方法与流程
背景技术:
1、本发明涵盖抑制造血祖细胞激酶1(hpk1)的一系列化合物。hpk1主要在造血细胞内表达,而且在t细胞和树突细胞(dc)中用作负调控因子。通过经基因敲入激酶死亡(kd)酶而消除hpk1的激酶活性,证明了其参与抗肿瘤免疫反应(hernandes等人,us2016/0158360)。在机理上,hpk1 kd降低了t细胞内下游蛋白slp76的磷酸化,从而增加了抗cd3和抗cd28刺激时促免疫细胞因子il-2和ifnγ的释放。在表达kd酶的小鼠肉瘤模型中,已观察到肿瘤微环境(tme)中肿瘤浸润淋巴细胞(til)的增加以及肿瘤生长的减少(liu等人,公共科学图书馆-综合(plos one),14卷(3期):e0212670)。此外,人们正在研究小分子hpk1抑制剂,而且其作为单一药剂或与抗pd1抗体(chen等人,美国癌症研究协会年会(aacr annualmeeting),2020年6月,4513号海报)及抗ctla-4抗体(ciccone等人,美国癌症研究协会年会,2020年6月,942号海报)联用后的抗肿瘤活性已在各种癌症模型中得到证明。
2、业已表明,hpk1 kd t细胞对前列腺素e2(pge2)和腺苷施加的免疫抑制作用具有抵抗力,从而为抑制pge2和腺苷途径的化合物的协同抗癌作用研究提供了依据。例如,hpk1抑制剂与ep4或a2a/b抑制剂的联用可在癌症的治疗上呈现出优异的治疗益处(liu等人,公共科学图书馆-综合(plos one),14卷(3期):e0212670)。
3、鉴于hpk1在调节免疫细胞活性方面的作用,小分子抑制剂可作为单一药剂或与现有免疫疗法、靶向疗法或抑制pge2或腺苷途径的化合物联合用于癌症的治疗。
技术实现思路
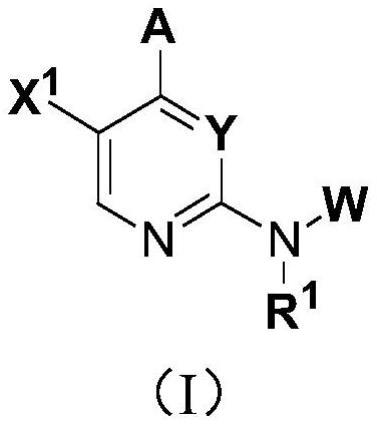
1、在一个方面,本发明提供式(i)杂芳基化合物或其药学上可接受的盐、酯或前体药物:
2、
3、其中,
4、y为n或cr23,其中,r23为h、卤素、烷基或取代烷基;
5、x1为h、卤素、氰基、烷基、环烷基、取代烷基、卤代烷基、烷氧基、烯基、烷硫基、羟基、氨基、氢羧基、烷氧基羰基、氨基羰基、芳基、杂芳基或取代或未取代的杂环烷基;或者,x1为稠合于式(i)中的6元杂芳基上的取代或未取代的杂芳基;
6、r1为h、烷基、环烷基、取代烷基或烯基;
7、r2和r3各自独立地是h、烷基、环烷基、取代烷基或烯基;或者,r2和r3连同它们所附接的原子一起形成环烷基;
8、a为任选取代的9元或10元稠合双环杂芳基或任选取代的5至7元芳基或杂芳基,其中,a可选地先与酰胺基、氨基、-nr-c(o)-、-c(o)-nr-、-r-c(o)-nr-或-c(o)-结合,然后连接至式(i)中的6元杂芳基,其中,a任选地被1~5个取代基取代,所述取代基各自独立地是烷基、卤代烷基、氰基烷基、环烷基、卤素、羟基、氰基、烷氧基、氨基或氨基羰基;其中,r为r10;
9、r10为h、烷基、取代烷基或环烷基;
10、w为5至7元单环、9至11元稠合双环或11至15元三环环烷基、杂环烷基、芳基或杂芳基,且w可任选地被1~5个取代基取代,所述取代基各自独立地是烷基、卤代烷基、卤素、羟基、氰基、烷氧基、环烷基、氨基羰基、杂环烷基、桥联双环环烷基或杂环烷基、螺环环烷基或杂环烷基;其中,每一取代基均可任选地被进一步取代;
11、其中,式i中的任何氢原子(h)可任选地被氘(d)取代,以使得各化合物具有更佳的药代动力学(pk)和/或效力。
12、在一些实施方式中,式(i)中的a为9至10元稠合双环杂芳基。
13、合适的a基团示例包括其中,
14、t1和t2各自独立地为n或cr5;
15、t3、t4和t5各自独立地为n、o、s、nr5或cr5;
16、t6和t7各自独立地为n或c;
17、r4和r5各自独立地在与碳连接时为h、烷基、环烷基、取代烷基、烯基、卤素或conr24r25;
18、r4和r5各自独立地在与氮原子连接时为h、烷基、取代烷基、烯基或conr24r2;
19、r6为h、卤素、烷基、取代烷基、烯基或nr26r27;
20、r7、r8及r9各自独立地为h、烷基、环烷基、取代烷基、烯基、卤素或conr24r25;
21、r24和r25各自独立地为h、烷基、取代烷基;或者,r24和r25与其所附接的原子一起形成杂环烷基;
22、r26和r27各自独立地为h、烷基或取代烷基;或者,r26和r27与其所附接的原子一起形成杂环烷基。
23、在一些其他实施方式中,式(i)中的a为具体的a示例包括
24、及其中,z为o或s。
25、在一些其他实施方式中,式(i)中的a为与酰胺基、氨基、-nr-c(o)-、-c(o)-nr-或-c(o)-结合的任选取代的5至7元芳基或杂芳基。
26、此类a的示例包括其中,
27、q为cr12或n;
28、r10为h、烷基、取代烷基或环烷基;
29、r11为卤素、氰基、烷基、取代烷基、烷氧基、亚烷基、s-烷基、羟基或nr2r3;
30、r12为h、卤素、氰基、烷基、取代烷基、烷氧基、亚烷基、s-烷基、羟基或nr2r3;而且
31、x2为h、卤素、氰基、烷基、取代烷基、烷氧基、亚烷基、s-烷基、羟基、nr2r3或cf3。
32、适合于本发明式(i)化合物的具体示例包括:
33、其中,r11为h、oh或cl;r22为h、f或cl;r33为h或ch3。
34、在一些实施方式中,式(i)中的w为取代芳基,取代杂芳基或杂环基。合适的w的示例包括
35、其中,
36、t8、t9、t10、t12和t13各自独立地选自n或cr22;
37、n和i各自独立地为1、2或3;
38、u为cr14r15或nr16;
39、r14、r15、r16、和r17各自独立地为h、烷基、环烷基、取代烷基、烯基、so2r2或co(nr2r3);
40、r18、r19、r20、和r21各自独立地为h、烷基、环烷基、取代烷基、烯基、卤素或co(nr2r3);
41、r22和r30各自独立地为h、卤素、羟基、氰基、烷基、环烷基、取代烷基、烷氧基、烯基、杂环烷基、cooh或co(nr2r3);
42、r13为h、烷基、取代烷基、环烷基、
43、l和r31各自独立地为n或cr22;
44、d为n或cr23;
45、e为cr24r25、nr26、o或so2;
46、r23、r24、r25、和r26各自独立地为h、羟基、卤素、烷基、环烷基、取代烷基、烯基、nr2r3或co(nr2r3);
47、r28、r29、r32和r33各自独立地为h、羟基、卤素、任选取代的烷基、任选取代的环烷基、任选取代的烯基或co(nr2r3);或者,r28及r29,或r32及r33,与其所连接的原子共同形成羰基;
48、p和q各自独立地为0、1、2或3。
49、式(i)中合适的w的其他示例包括其中,y为5或6元的任选取代的芳基或杂芳基。
50、在一些其他实施方式中,式(i)中的y为取代芳基或杂芳基。
51、在又一些其他实施方式中,式(i)中的w为并且在w中,t8、t9、t10、和t11均各自独立地为n或cr22。合适的w的示例包括单环或双环环烷基、芳基及杂芳基,并且w可任选地被1~4个取代基取代,所述取代基各自独立地为卤素、烷基、烷氧基、羟基、氰基、氨基羰基或杂环烷基。式(i)中合适的w的其他示例包括苯基、二氢萘啶基、哒嗪基、吡啶基、哌啶基、嘧啶基或吡唑基,其任选地被1~3个取代基取代,所述取代基各自独立地为卤素、烷基、卤代烷基、羟基、烷氧基、氰基、哌嗪基、烷基、哌啶基、吗啉基、氨基羰基或二氧代硫代吗啉基,其中所述哌嗪基可进一步由1~3个烷基取代。
52、合适的式(i)中w的具体示例包括
53、及
54、在一些实施方式中,式(i)中的x1为h、卤素、烷基、卤代烷基、烷氧基、烷硫基、氰基、羟基羰基、氨基羰基、杂芳基、环烷基或杂环烷基,其中的每一者均各自任选地被1~3个取代基取代(化学上允许时),所述取代基各自独立地为卤素或c1~3烷基。合适的x1示例包括cl、br、甲基、甲氧基、甲硫基、2,2,2-三氟乙基、二氟甲基、氰基、-co-nh2、乙烯基、环丙基及
55、在一些其他实施方式中,式(i)中的r1为h、甲基或卤代甲基。
56、式(i)化合物的示例包括:
57、
58、
59、
60、
61、
62、
63、
64、
65、
66、
67、
68、
69、
70、本发明化合物的其他示例包括如下:
71、
72、
73、在一些其他实施方式中,优选化合物选自如下:
74、
75、
76、
77、本发明的另一方面包括药物组合物,每种药物组合物均包括上述化合物以及药学上可接受的载体或赋形剂。此类药物组合物可包括第二治疗剂,该第二治疗剂可例如为免疫检查点抑制剂,或pge2或腺苷途径抑制剂。合适的免疫检查点抑制剂示例包括伊匹单抗(ipilimumab)、纳武单抗(nivolumab)以及派姆单抗(pembroluzimab)。
78、本发明的又一方面提供一种治疗需治疗对象的hpk1介导病症的方法。该方法包括:向所述对象施用治疗有效量的上述化合物或药物组合物。在一些实施方式中,所述病症为癌症(如乳腺癌、结肠直肠癌、肺癌、卵巢癌或胰腺癌)。
79、定义
80、除非上下文另有所指,否则本文所有部分(包括用途、方法及本发明的其他方面)中提到的式(i)涵盖本文中描述的所有其他分式、分组、优选项、实施方式及实施例。
81、除非另有说明,否则说明书及权利要求书中使用的下列术语具有下述含义。
82、“烷基”是指饱和直链烃或饱和分支烃。烷基的示例包括甲基、乙基、丙基、2-丙基、正丁基、异丁基、叔丁基、戊基、己基等,优选甲基、乙基、丙基或2-丙基。代表性的饱和直链烷基包括甲基、乙基、正丙基、正丁基、正戊基、正己基等,而饱和分支烷基包括异丙基、仲丁基、异丁基、叔丁基、异戊基等。在本文中,成环的烷基称为“环烷基”。“c0~4烷基”是指具有0个、1个、2个、3个或4个碳原子的烷基。具有0个碳原子的c0~4烷基在处于末端的情况下指氢原子,在中连情况下指直连键。烷基可通过一个或两个连接点与分子连接。
83、“取代烷基”是指以一个或多个卤素、羟基、氰基、烷氧基、氨基、甲基氨基、二甲基氨基、砜基、磺酰胺基、芳基、杂芳基及杂环基取代的烷基。取代烷基的示例包括三氟乙基、羟基乙基、氰基乙基、甲氧基乙基及三氟丙基。除非另外明确指定数目,否则卤代烷基可包括化学上允许的最大数目个卤素原子,以作为其烷基的取代基。氟代乙基可例如为-ch2cf3、-chf-ch3或-ch2ch2f。
84、“烷氧基”是指与氧原子连接的饱和直链烃或饱和分支烃。烷氧基的示例包括甲氧基、乙氧基、丙氧基、2-丙氧基、正丁氧基、异丁氧基、叔丁氧基、戊氧基、己氧基等,优选甲氧基、乙氧基、丙氧基或2-丙氧基。代表性的饱和直链烷氧基包括甲氧基、乙氧基、正丙氧基、正丁氧基、正戊氧基、正己氧基等,而饱和分支烷氧基包括异丙氧基、仲丁氧基、异丁氧基、叔丁氧基、异戊氧基等。在本文中,成环的烷氧基称为“环烷氧基”。“c1~4烷氧基”是指具有1个、2个、3个或4个碳原子的烷基。烷氧基可通过一个或两个连接点与分子连接。
85、“烯基”是指非饱和的直链烃或分支烃。烯基的示例包括乙烯基和丙烯基。烯基也可由一个或多个烷基取代。
86、“酰胺基”既包括-c(=o)nr2(即氨基羰基),也包括r-c(=o)-nr-,其中,以羰基或氨基作为键合原子。
87、“氨基”是指-nr'r”基团,其中,r'和r”可以相同,也可不同,而且均可为氢(对应于-nh2)、烷基(对应于-nh-烷基)或其他化学部分(如氰基或环烷基)。
88、“环烷基”是指饱和环烃。环烷基的示例包括环丙基、环丁基、环戊基及环己基。环烷基可通过一个或两个连接点与分子连接。环烷基可进而由一个或多个卤素可选取代。
89、“卤素”是指氟、氯、溴或碘。
90、“芳基”是指具有完全共轭π电子体系且由6至12个碳原子组成的全碳单环或稠环多环(即共享成对相邻碳原子的环)基团。芳基的非限制性示例为苯基、萘基及蒽基。
91、“杂芳基”是指具有完全共轭π电子体系且由5至12个成环原子组成的单环或稠环(即共享成对相邻碳原子的环),其中,成环原子当中含有一个、两个、三个或四个选自n、o或s的成环杂原子,其他成环原子为c。未取代杂芳基的非限制性示例为吡咯、呋喃、噻吩、咪唑、恶唑、噻唑、吡唑、吡啶、嘧啶、喹啉、异喹啉、嘌呤、三唑、四唑、三嗪、咔唑、苯并咪唑、苯并恶唑、苯并噻唑、吲唑及喹唑啉。杂芳基可以取代或不取代。
92、“杂环基”或“杂环烷基”是指具有3至14个成环原子的饱和、不饱和或芳环环系,其中,一个、两个或三个成环原子为选自n、o或s(o)m(其中,m为0~2的整数)的杂原子,其余成环原子为c,其中,一个或两个c原子可由羰基可选取代。除非另有说明,否则“杂环基”或“杂环烷基”一词(如“饱和杂环基”)涵盖杂芳基。杂环基可通过一个或两个连接点与分子连接。
93、取代基可以组合,但是只有在此类组合能够产生稳定或化学上允许(即在40℃或更低温度下保持至少一周时基本不变)的化合物才如此。
94、构成本发明化合物的各种官能团和取代基一般选择为使得本发明化合物的分子量不超过1000。更通常而言,该化合物的分子量小于750,例如,小于700,或小于650,或小于600,或小于550。更优选地,该分子量小于525,且例如为500或更小。
95、“可选”或者“任选”是指随后描述的事件或情形可发生,但可不必发生,而且该表述涵盖所述事件或情形会发生以及所述事件或情形不发生这两种状况。例如,“杂环基由烷基可选取代”是指,可存在这一烷基,但不一定非得存在该烷基,而且该表述涵盖所述杂环基由烷基取代以及所述杂环基未由烷基取代这两种情形。
96、通过加酸制备的“药学上可接受的盐”为由随后形成无毒酸根阴离子的酸形成的盐,如盐酸盐、氢溴酸盐、硫酸盐、磷酸盐或酸性磷酸盐、乙酸盐、马来酸盐、富马酸盐、乳酸盐、酒石酸盐、柠檬酸盐及葡萄糖酸盐。
97、当本发明化合物含有羧基时,该化合物可按照常规方法(如羧酸与醇的缩合反应),通过与相应的醇(如c1~6醇)反应而制成药学上可接受的酯。
98、“药物组合物”是指本文所述化合物当中的一种或多种或其药学上可接受的盐或前体药物与药学上可接受的赋形剂等其他化学成分的混合物。药物组合物的目的旨在有利于化合物施用至生物体。
99、“药学上可接受的赋形剂”是指添加至药物组合物中以进一步有利于化合物的施用的惰性物质。赋形剂的非限制性示例包括碳酸钙、磷酸钙、各种糖和各种类型的淀粉、纤维素衍生物、明胶、植物油及聚乙二醇。
100、“治疗有效量”是指在所施用的化合物在一定程度上缓解被治疗病症的一种或多种症状的量。在癌症治疗方面,治疗有效量是指具有如下作用的量:(1)减小肿瘤尺寸;(2)抑制肿瘤转移;(3)抑制肿瘤生长;以及/或者(4)缓解癌症相关的一种或多种症状。
101、化学基团中具有自由基且与另一原子键合的原子以*,或波浪线表示,例如,见
102、当取代基未与特定原子键合时(例如,如所示),该取代基可按照化学上允许的方式与任何成环原子(碳或氮)键合。
103、实施例1:
104、5-氯-4-(1-甲基-1h-苯并[d]咪唑-6-基)-n-(4-(4-甲基哌嗪-1-基)苯基)嘧啶-2-胺
105、
106、路线图
107、
108、步骤1:1-甲基-6-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1h-苯并[d]咪唑
109、
110、在混于二恶烷(250ml)内的6-溴-1-甲基-1h-苯并[d]咪唑化合物(10.0g,47.21mmol,1.0当量)、双(频哪醇合)二硼(b2pin2)
111、(14.43g,56.21mmol,1.2当量)及乙酸钾(9.29g,94.11mmol,3.0当量)中,加入[1,1'-双(二苯基膦)二茂铁]二氯化钯(pd(dppf)cl2)(1.0g,1.36mmol,0.03当量)。混合物以氮气脱气3次,并在氮气气氛中100℃搅拌10小时。反应混合物的液相色谱质谱联用(lc-ms)分析表明完全转化至所需产物。混合物经乙酸乙酯稀释(200ml×2)、饱和氯化钠水溶液洗涤(150ml)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(二氯甲烷:甲醇=0~2%),获得灰色固体的1-甲基-6-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1h-苯并[d]咪唑(10.1g,82.5%)。
112、lc-ms m/z:259.1(m+1)+。
113、步骤2:6-(2,5-二氯嘧啶-4-基)-1-甲基-1h-苯并[d]咪唑
114、
115、在混于二恶烷/水(100ml/50ml)内的1-甲基-6-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1h-苯并[d]咪唑化合物(7.0g,27.13mmol,1.0当量)及磷酸钾(11.5g,54.26mmol,2.0当量)中,加入2,4,5-三氯嘧啶(5.97g,32.56mmol,1.2当量)及四(三苯基膦)钯(pd(pph3)4)(1g,0.865mol,0.03当量)。混合物以氮气脱气3次,并在氮气气氛中80℃搅拌10小时。lc-ms表明反应完成。混合物经减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(石油醚:乙酸乙酯=65:35,二氯甲烷:甲醇=100:0~97:3),获得淡黄色固体的6-(2,5-二氯嘧啶-4-基)-1-甲基-1h-苯并[d]咪唑(3.4g,44.9%)。
116、lc-ms m/z:279.01(m+1)+。
117、步骤3:5-氯-4-(1-甲基-1h-苯并[d]咪唑-6-基)-n-(4-(4-甲基哌嗪-1-基)苯基)嘧啶-2-胺
118、
119、在混于无水二恶烷(15ml)内的6-(2,5-二氯嘧啶-4-基)-1-甲基-1h-苯并[d]咪唑化合物(200mg,0.719mmol,1.0当量)、4-(4-甲基哌嗪-1-基)苯胺化合物(137.60mg,0.719mmol,1.0当量)及碳酸铯(703mg,2.158mmol,3.0当量)中,加入[(4,5-双(二苯基膦)-9,9-二甲基氧杂蒽)-2-(2'-氨基-1,1'-联苯)]甲烷磺酸钯(ii)(xantphos-pd-g3)(80mg,0.084mol,0.11当量)。混合物以氮气脱气3次,并在氮气气氛中110℃搅拌16小时。lc-ms表明反应完成。混合物经过滤及减压浓缩,获得粗产物。粗产物经制备型hplc纯化,获得黄色固体的实施例1(42.0mg,5.6%,甲酸盐)。
120、制备型hplc条件:
121、方法a:仪器:岛津(shimadzu)lc8ap制备型hplc系统;色谱柱:boston ods(250mm×21.2mm×10μm,);流动相:溶有0.1%甲酸的水/乙腈;梯度:乙腈在1分钟至5分钟时间段为5%,在5分钟至20分钟时间段为5%至25%,在20分钟至25分钟时间段为25%;柱温:25℃;检测波长:214/254nm;流速:20ml/min;
122、lc-ms m/z:434.5(m+1)+;
123、1h nmr(400mhz,d6-二甲亚砜(dmso)):δ9.65(s,1h),8.51(s,1h),8.29(s,1h),8.14(s,1h),7.98(s,1h),7.73
124、(d,j=8.2hz,1h),7.63(d,j=8.1hz,1h),7.55(d,j=8.4hz,2h),6.85(d,j=8.4hz,2h),3.86(s,3h),3.05~3.02(m,4h),2.44~2.40(m,4h),2.19(s,3h)。
125、
126、
127、实施例4:
128、5-氯-4-(1-甲基-1h-苯并[d]咪唑-6-基)-n-(4-(4-甲基哌嗪-1-基)苯基)嘧啶-2-胺
129、
130、路线图
131、
132、步骤1:1-甲基-5-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1h-苯并[d]咪唑
133、
134、在混于二恶烷(20ml)内的5-溴-1-甲基-1h-苯并[d]咪唑化合物(5.0g,23.8mmol,1.0当量)、b2pin2(7.25g,28.56mmol,1.2当量)及乙酸钾(4.66g,47.6mmol,2.0当量)中,加入pd(dppf)cl2(980.56mg,1.19mol,0.05当量)。混合物以氮气脱气3次,并在氮气气氛中80℃搅拌3小时。反应混合物的lcms分析表明反应完成。混合物进行过滤和浓缩,残余物经乙酸乙酯稀释(60ml)、饱和氯化钠水溶液洗涤(50ml)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(二氯甲烷:甲醇=100:0),获得白色固体的1-甲基-5-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1h-苯并[d]咪唑(5.3g,86.8%)。
135、lc-ms m/z:259.3(m+1)+。
136、步骤2:5-(2,5-二氯嘧啶-4-基)-1-甲基-1h-苯并[d]咪唑
137、
138、在混于四氢呋喃(thf)(50ml)和水(50ml)内的1-甲基-5-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1h-苯并[d]咪唑化合物(5.3g,20.5mmol,1.1当量)、2,4,5-三氯嘧啶(3.4g,18.7mmol,1.0当量)及碳酸钠(5.9g,56.1mmol,3.0当量)中,加入乙酸钯(209mg,1.87mol,0.1当量)和三苯基膦(pph3)(1.9g,7.48mol,0.4当量)。混合物以氮气脱气3次,并在氮气气氛中回流搅拌3小时。lcms表明反应完成。混合物经乙酸乙酯稀释(40ml)、饱和氯化钠水溶液洗涤(40ml×3)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(二氯甲烷:甲醇=98:2),获得白色固体的5-(2,5-二氯嘧啶-4-基)-1-甲基-1h-苯并[d]咪唑(1.5g,22.7%)。
139、lc-ms m/z:279.3(m+1)+。
140、步骤3:5-氯-4-(1-甲基-1h-苯并[d]咪唑-6-基)-n-(4-(4-甲基哌嗪-1-基)苯基)嘧啶-2-胺
141、
142、在混于异丙醇(2ml)内的5-(2,5-二氯嘧啶-4-基)-1-甲基-1h-苯并[d]咪唑化合物(100mg,0.36mmol,1.0当量)及4-(4-甲基哌嗪-1-基)苯胺化合物(68mg,0.36mmol,1.0当量)中,加入三氟乙酸(tfa)(0.5ml)。混合物以氮气脱气3次,并在氮气气氛中于密封管内110℃搅拌16小时。反应混合物的薄层色谱(tlc)分析表明反应完成。混合物溶于乙酸乙酯(10ml),并经饱和碳酸氢钠水溶液洗涤(5ml×3)、无水硫酸钠干燥、过滤以及减压浓缩,获得粗产物。粗产物经制备型hplc纯化,获得黄色固体的实施例4化合物(28.6mg,19.3%)。
143、制备型hplc条件:
144、方法a:岛津lc8ap制备型hplc系统;色谱柱:boston ods(250mm×21.2mm×10μm,);流动相:溶有0.1%甲酸的水/乙腈;梯度:乙腈在1分钟至5分钟时间段为5%,在5分钟至25分钟时间段为5%至25%,在25分钟至30分钟时间段为25%;柱温:25℃;检测波长:214/254nm;流速:20ml/min;
145、lc-ms m/z:434.2(m+1)+;
146、1h nmr(400mhz,d6-dmso)δ9.68(s,1h),8.52(s,1h),8.29(s,1h),8.12(s,1h),8.09(s,1h),7.70(d,j=5.6hz,2h),7.58(d,j=8.8hz,2h),6.89(d,j=8.8hz,2h),3.87(s,3h),3.15(t,j=7.6hz,4h),2.83(t,j=7.6hz,4h),2.47(s,3h)。
147、
148、
149、
150、
151、
152、实施例16:
153、n-(5-氟-4-(1-甲基-1h-苯并[d]咪唑-5-基)嘧啶-2-基)-6-甲基-5,6,7,8-四氢-1,6-萘啶-3-胺
154、
155、路线图
156、
157、步骤1:5-(2-氯-5-氟嘧啶-4-基)-1-甲基-1h-苯并[d]咪唑
158、
159、在混于二恶烷/水(8ml/2ml)内的2,4-二氯-5-氟嘧啶化合物(194mg,1.16mmol,1.0当量)、(1-甲基-1h-苯并[d]咪唑-5-基)硼酸化合物(300mg,1.16mmol,1.0当量)、pph3(60mg,0.23mmol,0.2当量)、碳酸钠(370mg,3.48mmol,3.0当量)中,加入乙酸钯(26mg,0.12mmol,0.1当量)。混合物在氮气气氛中110℃搅拌14小时。反应混合物的lcms分析表明反应完成。混合物经乙酸乙酯稀释(10ml)、饱和氯化钠水溶液洗涤(5ml)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(二氯甲烷:甲醇=0~1%),获得浅白色固体的5-(2-氯-5-氟嘧啶-4-基)-1-甲基-1h-苯并[d]咪唑化合物(100mg,33%)。
160、lc-ms m/z:263.3(m+1)+。
161、步骤2:n-(5-氟-4-(1-甲基-1h-苯并[d]咪唑-5-基)嘧啶-2-基)-6-甲基-5,6,7,8-四氢-1,6-萘啶-3-胺
162、
163、在混于二恶烷(5ml)内的5-(2-氯-5-氟嘧啶-4-基)-1-甲基-1h-苯并[d]咪唑化合物(100mg,0.38mmol,1.0当量)、6-甲基-5,6,7,8-四氢-1,6-萘啶-3-胺化合物(82mg,0.45mmol,1.2当量)、碳酸铯(410mg,1.26mmol,3.0当量)中,加入xantphos-pd-g3(20mg,0.02mmol,0.05当量)。混合物在氮气气氛中110℃搅拌0.5小时。反应混合物的lcms分析表明反应完成。混合物经乙酸乙酯稀释(10ml)、饱和氯化钠水溶液洗涤(5ml)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经制备型hplc纯化,获得黄色固体的实施例16(26.6mg,26%)。制备型hplc条件:
164、方法a:仪器:岛津lc8ap制备型hplc系统;色谱柱:boston ods(250mm×21.2mm×10μm,);流动相:溶有0.1%甲酸的水/乙腈;梯度:乙腈在1分钟至5分钟时间段为5%,在5分钟至20分钟时间段为5%至25%,在20分钟至25分钟时间段为25%;柱温:25℃;检测波长:214/254nm;流速:20ml/min;
165、lc-ms m/z:390.4(m+1)+;
166、1h nmr(400mhz,cd3od):δ8.67(s,1h),8.46~8.37(m,3h),8.23(s,1h),8.17(s,1h),8.12(d,j=8.4,1h),7.66(d,j=8.8,1h),4.03(s,2h),3.93(s,3h),3.20(t,j=6.0,2h),3.08(t,j=5.6,2h),2.75(s,3h)。
167、
168、
169、实施例19:
170、5-氯-4-(1-甲基-1h-苯并[d]咪唑-5-基)-n-(4-(哌嗪-1-基)苯基)嘧啶-2-胺
171、
172、路线图
173、
174、步骤1:4-(4-((5-氯-4-(1-甲基-1h-苯并[d]咪唑-5-基)嘧啶-2-基)氨基)苯基)哌嗪-1-羧酸叔丁酯
175、
176、在混于二恶烷(10ml)内的5-(2,5-二氯嘧啶-4-基)-1-甲基-1h-苯并[d]咪唑化合物(200mg,0.72mmol,1.0当量)、4-(4-氨基苯基)哌嗪-1-羧酸叔丁酯化合物(200mg,0.72mmol,1.0当量)及碳酸铯(0.65g,2mmol,2.8当量)中,加入xantphos-pd-g3(50mg,0.05mol,0.07当量)。混合物以氮气脱气3次,并在氮气气氛中110℃搅拌16小时。反应混合物的lcms分析表明反应完成。混合物以硅藻土过滤,滤过物经减压浓缩,获得棕色固体的4-(4-((5-氯-4-(1-甲基-1h-苯并[d]咪唑-5-基)嘧啶-2-基)氨基)苯基)哌嗪-1-羧酸叔丁酯(300mg,77.1%)粗产物。
177、lc-ms m/z:521.2(m+1)+。
178、步骤2:5-氯-4-(1-甲基-1h-苯并[d]咪唑-5-基)-n-(4-(哌嗪-1-基)苯基)嘧啶-2-胺
179、
180、在混于二氯甲烷(3ml)内的4-(4-((5-氯-4-(1-甲基-1h-苯并[d]咪唑-5-基)嘧啶-2-基)氨基)苯基)哌嗪-1-羧酸叔丁酯化合物(粗产物,300mg,0.55mmol,1.0当量)中,加入tfa(3ml)。混合物在氮气气氛中25℃搅拌1小时。反应混合物的lcms分析表明反应完成。混合物经减压浓缩,获得粗产物。粗产物经制备型hplc纯化,获得棕色固体的实施例19化合物(24.8mg,8.2%)。
181、制备型hplc条件:
182、方法c:仪器:岛津lc8ap制备型hplc系统;色谱柱:boston ods(250mm×21.2mm×10μm,);流动相:溶于水/乙腈的0.1%tfa;梯度:乙腈在1分钟至5分钟时间段为5%,在5分钟至20分钟时间段为20%至40%,在20分钟至25分钟时间段为40%;柱温:25℃;检测波长:214/254nm;流速:20ml/min;
183、lc-ms m/z:420.3(m+1)+;
184、1h nmr(400mhz,cd3od):δ9.19(s,1h),8.46(s,1h),8.30(s,1h),8.09(d,j=8.4hz,1h),7.95(d,j=8.4hz,1h),7.61(d,j=8.4hz,2h),6.97(d,j=8.4hz,2h),4.12(s,3h),3.33(s,4h),3.28(s,4h)。
185、实施例20:
186、n-(4-(1h-苯并[d]咪唑-5-基)-5-氯嘧啶-2-基)-6-甲基-5,6,7,8-四氢-1,6-萘啶-3-胺
187、
188、路线图
189、
190、步骤1:5-溴-1-((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑
191、
192、在混于thf内的5-溴-1h-苯并[d]咪唑化合物(2.0g,10.1mmol,1.0当量)中,加入氢化钠(487mg,以60%散于矿物油中,12.1mmol,1.2当量)。混合物以氮气脱气3次,并在氮气气氛中0℃搅拌0.5小时,随后加入2-(三甲基硅烷基)乙氧甲基氯(semcl)(2.5g,15.2mmol,1.5当量),并室温搅拌3小时。反应混合物的lcms分析表明完全转化至所需产物。反应溶液以氯化铵淬灭,混合物经减压浓缩,去除溶剂。残余物溶于乙酸乙酯(60ml)中,并经饱和氯化钠水溶液洗涤(50ml×2)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(二氯甲烷:甲醇=100:0~99:1),获得无色清油的5-溴-1-((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑化合物(2.3g,52.1%)。
193、lc-ms m/z:327.1(m+1)+。
194、步骤2:5-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1-((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑
195、
196、在混于dmso(20ml)内的5-溴-1-((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑化合物(1.3g,4.0mmol,1.0当量)、b2pin2(1.52g,6.0mmol,1.50当量)及乙酸钾(784mg,8.0mmol,2.0当量)中,加入pd(dppf)cl2(164mg,0.2mol,0.05当量)。混合物以氮气脱气3次,并在氮气气氛中80℃搅拌2小时。反应混合物的lcms分析表明反应完成。混合物经乙酸乙酯稀释(15ml)、饱和氯化钠水溶液洗涤(5ml)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(二氯甲烷:甲醇=0~1%),获得淡黄色固体的5-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1-((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑化合物(600mg,37.3%)。
197、lc-ms m/z:385(m+1)+。
198、步骤3:5-(2,5-二氯嘧啶-4-基)-1-((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑
199、
200、在混于无水thf/水(10ml/5ml)内的5-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1-((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑化合物(600mg,1.6mmol,1.0当量)、2,4,5-三氯嘧啶化合物(293mg,1.6mmol,1.0当量)、碳酸钠(508.8mg,4.8mmol,3.0当量)及pph3(191mg,0.784mmol,0.4当量)中,加入乙酸钯(311mg,0.187mol,0.11当量)。混合物以氮气脱气3次,并在氮气气氛中回流搅拌3小时。反应混合物的lcms分析表明完全转化至所需产物。混合物经减压浓缩,去除溶剂。残余物溶于二氯甲烷(30ml)中,并经饱和氯化钠水溶液洗涤(15ml×2)、无水硫酸钠干燥、过滤及减压浓缩获得粗产物。粗产物经硅胶柱层析纯化(二氯甲烷:甲醇=0~5%),获得淡黄色固体的5-(2,5-二氯嘧啶-4-基)-1-((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑化合物(180mg,33.5%)。
201、lc-ms m/z:395.2(m+1)+。
202、步骤4:n-(5-氯-4-(1-((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑-5-基)嘧啶-2-基)-6-甲基-5,6,7,8-四氢-1,6-萘啶-3-胺
203、
204、在混于二恶烷(3ml)内的5-(2,5-二氯嘧啶-4-基)-1-((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑化合物(170mg,0.43mmol,1.00当量)、6-甲基-5,6,7,8-四氢-1,6-萘啶-3-胺化合物(70mg,0.43mmol,1.50当量)及碳酸铯(420mg,1.29mmol,3.00当量)中,加入xanphos-pd-g3(20mg,0.02mol,0.05当量)。混合物以氮气脱气3次,并在110℃下微波搅拌0.5小时。反应混合物的lcms分析表明反应完成。混合物经乙酸乙酯稀释(15ml)、饱和氯化钠水溶液洗涤(5ml)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(二氯甲烷:甲醇=0~4%),获得淡黄色固体的n-(5-氯-4-(1-((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑-5-基)嘧啶-2-基)-6-甲基-5,6,7,8-四氢-1,6-萘啶-3-胺化合物(150mg,83.3%)。
205、lc-ms m/z:522.2(m+1)+。
206、步骤5:n-(4-(1h-苯并[d]咪唑-5-基)-5-氯嘧啶-2-基)-6-甲基-5,6,7,8-四氢-1,6-萘啶-3-胺
207、
208、在混于二氯甲烷(dcm)(5ml)内的n-(5-氯-4-(1-((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑-5-基)嘧啶-2-基)-6-甲基-5,6,7,8-四氢-1,6-萘啶-3-胺化合物(150mg,0.287mmol,1.0当量)中,加入tfa(5ml)。混合物在氮气气氛中室温搅拌16小时。反应混合物的lcms分析表明完全转化至所需产物。混合物经减压浓缩,去除溶剂,获得粗产物。粗产物经制备型hplc纯化,获得白色固体的实施例20化合物(12.2mg,26%)。
209、制备型hplc条件:
210、方法c:仪器:岛津lc8ap制备型hplc系统;色谱柱:boston ods(250mm×21.2mm×10μm,);流动相:溶于水/乙腈的0.1%tfa;梯度:乙腈在1分钟至5分钟时间段为5%,在5分钟至20分钟时间段为20%至40%,在20分钟至25分钟时间段为40%;柱温:25℃;检测波长:214/254nm;流速:20ml/min;
211、lc-ms m/z:392.4(m+1)+;
212、1h nmr(400mhz,cd3od):δ8.71(s,1h),8.48(s,1h),8.34(s,1h),8.28(s,1h),8.16(d,j=14.9,2h),7.81(d,j=8.4,1h),7.69(d,j=8.4,1h),4.07(s,2h),3.28~3.26(t,j=6.0,2h),3.10(t,j=6.0,2h),2.78(s,3h)。
213、
214、
215、实施例22:
216、n-(4-(1h-苯并[d]咪唑-5-基)-5-氯嘧啶-2-基)-6-甲基-5,6,7,8-四氢-1,6-萘啶-3-胺
217、
218、路线图
219、
220、步骤1:5-溴-2-(溴甲基)烟酸乙酯
221、
222、在混于四氯化碳(60ml)内的5-溴-2-甲基烟酸乙酯化合物(5g,20.49mmol,1.0当量)及n-溴代丁二酰亚胺(nbs)(3.68g,20.69mmol,1.01当量)中,加入偶氮二异丁腈(aibn)(3.36g,20.49mol,1.0当量)。混合物以氮气脱气3次,并在氮气气氛中回流搅拌16小时。反应混合物的lcms分析表明完全转化至所需产物。混合物过滤后,滤过物溶于二氯甲烷(50ml)中,并经饱和氯化钠水溶液洗涤(50ml×3)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(石油醚:乙酸乙酯=85:15),获得黄色固体的5-溴-2-(溴甲基)烟酸乙酯化合物(3g,33.3%)。
223、lc-ms m/z:324.2(m+1)+。
224、步骤2:3-溴-6-甲基-6,7-二氢-5h-吡咯并[3,4-b]吡啶-5-酮
225、
226、在悬于甲醇(30ml)内的5-溴-2-(溴甲基)烟酸乙酯化合物(2.8g,8.66mmol,1.0当量)中,加入甲胺(50ml),并在氮气气氛中25℃搅拌20小时。反应混合物的lcms分析表明反应完成。混合物经减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(石油醚:乙酸乙酯=85:15),获得白色固体的3-溴-6-甲基-6,7-二氢-5h-吡咯并[3,4-b]吡啶-5-酮化合物(1.2g,60.9%)。
227、lc-ms m/z:227.3(m+1)+。
228、步骤3:3-溴-6,7,7-三甲基-6,7-二氢-5h-吡咯并[3,4-b]吡啶-5-酮
229、
230、0℃下,在混于无水thf(10ml)内的3-溴-6-甲基-6,7-二氢-5h-吡咯并[3,4-b]吡啶-5-酮化合物(900mg,3.96mmol,1.0当量)中,加入氢化钠(495mg,11.88mol,3.0当量)。混合物在氮气气氛中0℃搅拌0.5小时后,加入碘甲烷(1.68g,11.88mmol,3.0当量),并在氮气气氛中继续70℃搅拌4小时。反应混合物的lcms分析表明完全转化至所需产物。反应物以氯化铵水溶液淬灭,残余物溶于乙酸乙酯(10ml)中,并经饱和氯化钠水溶液洗涤(20ml×3)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(二氯甲烷:甲醇=98:2),获得白色固体的标题所示化合物(380mg,37.5%)。
231、lc-ms m/z:255.3(m+1)+。
232、步骤4:3-((二苯基亚甲基)氨基)-6,7,7-三甲基-6,7-二氢-5h-吡咯并[3,4-b]吡啶-5-酮
233、
234、在混于二恶烷(5ml)内的3-溴-6,7,7-三甲基-6,7-二氢-5h-吡咯并[3,4-b]吡啶-5-酮化合物(380mg,1.49mmol,1.0当量)及二苯甲酮亚胺化合物(406mg,2.24mmol,1.5当量)中,加入4,5-双二苯基膦-9,9-二甲基氧杂蒽(xantphos)(173mg,0.29mmol,0.2当量)、碳酸铯(975mg,2.99mmol,2.0当量)、及三(二亚苄基丙酮)二钯(pd2(dba)3)(137mg,0.14mmol,0.1当量)。混合物以氮气脱气3次,并在氮气气氛中110℃搅拌16小时。反应混合物的lcms分析表明完全转化至所需产物。混合物经乙酸乙酯稀释(5ml)、饱和氯化钠水溶液洗涤(5ml×2)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物为白色固体(400mg,75.3%),直接用于下一步骤的反应。
235、lc-ms m/z:356.4(m+1)+。
236、步骤5:3-氨基-6,7,7-三甲基-6,7-二氢-5h-吡咯并[3,4-b]吡啶-5-酮
237、
238、在混于二恶烷(5ml)内的3-((二苯基亚甲基)氨基)-6,7,7-三甲基-6,7-二氢-5h-吡咯并[3,4-b]吡啶-5-酮化合物(400mg,1.12mmol,1.0当量)中,加入盐酸(2.5ml,2n)。混合物以氮气脱气3次,并在氮气气氛中25℃搅拌3小时。反应混合物的lcms分析表明完全转化至所需产物。混合物经乙酸乙酯稀释(5ml)、饱和碳酸氢钠水溶液洗涤(5ml×3)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(二氯甲烷:甲醇=0~3%),获得棕色固体的标题所示化合物(240mg,100%)。
239、lc-ms m/z:192.2(m+1)+。
240、步骤6:3-氨基-6,7,7-三甲基-6,7-二氢-5h-吡咯并[3,4-b]吡啶-5-酮
241、
242、在混于二恶烷(4ml)内的3-氨基-6,7,7-三甲基-6,7-二氢-5h-吡咯并[3,4-b]吡啶-5-酮化合物(100mg,0.52mmol,1.0当量)、6-(2,5-二氯嘧啶-4-基)-1-甲基-1h-苯并[d]咪唑化合物(175mg,0.62mmol,1.2当量)及碳酸铯(500mg,1.56mmol,3.0当量)中,加入xantphos-pd-g3(50mg,0.05mol,0.1当量)。混合物以氮气脱气3次,并在氮气气氛中110℃搅拌16小时。反应混合物的lcms分析表明反应完成。混合物经乙酸乙酯稀释(15ml)、饱和氯化钠水溶液洗涤(5ml)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经制备型hplc纯化,获得白色固体的实施例22(7.0mg,3.1%)。
243、制备型hplc条件:
244、方法c:仪器:岛津lc8ap制备型hplc系统;色谱柱:boston ods(250mm×21.2mm×10μm,);流动相:溶有0.1%甲酸的水/乙腈;梯度:乙腈在1分钟至5分钟时间段为5%,在5分钟至20分钟时间段为20%至40%,在20分钟至25分钟时间段为40%;柱温:25℃;检测波长:214/254nm;流速:20ml/min;
245、lc-ms m/z:434.2(m+1)+;
246、1h nmr(400mhz,cd3od):δ9.27(s,1h),8.85(d,j=11.2hz,2h),8.61(s,1h),8.41(s,1h),8.15(d,j=8.0hz,1h)7.95(d,j=12.0hz,1h),4.19(s,3h),3.06(s,3h),1.50(s,6h)。
247、
248、
249、实施例24:
250、n-(5-氯-4-(1-甲基-1h-苯并[d]咪唑-5-基)嘧啶-2-基)-5,6,7,8-四氢-1,6-萘啶-3-胺
251、
252、路线图
253、
254、步骤1:3-((5-氯-4-(1-甲基-1h-苯并[d]咪唑-5-基)嘧啶-2-基)氨基)-7,8-二氢-1,6-萘啶-6(5h)-羧酸叔丁酯
255、
256、在混于无水二恶烷(4ml)内的5-(2,5-二氯嘧啶-4-基)-1-甲基-1h-苯并[d]咪唑化合物(100mg,0.35mmol,1.0当量)、3-氨基-7,8-二氢-1,6-萘啶-6(5h)-羧酸叔丁酯化合物(130mg,0.53mmol,1.5当量)及碳酸铯(350mg,1.05mmol,3当量)中,加入xantphos-pd-g3(35mg,0.03mol,0.1当量)。混合物以氮气脱气3次,并在氮气气氛中110℃搅拌16小时。反应混合物的lcms分析表明完全转化至所需产物。混合物过滤后,经减压浓缩,去除溶剂。残余物(200mg,100%)直接用于下一步骤的反应。
257、lc-ms m/z:492.3(m+1)+。
258、步骤2:3-((5-氯-4-(1-甲基-1h-苯并[d]咪唑-5-基)嘧啶-2-基)氨基)-7,8-二氢-1,6-萘啶-6(5h)-羧酸叔丁酯
259、
260、在悬于dcm(2ml)内的3-((5-氯-4-(1-甲基-1h-苯并[d]咪唑-5-基)嘧啶-2-基)氨基)-7,8-二氢-1,6-萘啶-6(5h)-羧酸叔丁酯化合物(200mg,0.40mmol,1.0当量)中,加入tfa(2ml),并在氮气气氛中25℃搅拌4小时。反应混合物的lcms分析表明反应完成。混合物经减压浓缩,获得粗产物。粗产物溶于dmf(3ml)中,并经制备型hplc纯化,获得黄色固体的实施例24(64.0mg,40.2%)。
261、制备型hplc条件:
262、方法a:仪器:岛津lc8ap制备型hplc系统;色谱柱:boston ods(250mm×21.2mm×10μm,);流动相:溶于水/乙腈的0.1% tfa;梯度:乙腈在1分钟至5分钟时间段为5%,在5分钟至20分钟时间段为5%至25%,在20分钟至25分钟时间段为25%;柱温:25℃;检测波长:214/254nm;流速:20ml/min;
263、lc-ms m/z:392.1(m+1)+;
264、1h nmr(400mhz,cd3od):δ9.36(s,1h),9.01(s,1h),8.63(s,1h),8.35(s,1h),8.26(s,1h),8.14(s,1h),8.03(s,1h),4.45(s,2h),4.16(s,3h),3.62(s,2h),3.27~3.22(m,2h)。
265、实施例25:
266、5-甲氧基-4-(1-甲基-1h-苯并[d]咪唑-5-基)-n-(4-(4-甲基哌嗪-1-基)苯基)嘧啶-2-胺
267、
268、实施例26:
269、4-(1-甲基-1h-苯并[d]咪唑-5-基)-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-5-醇
270、
271、路线图
272、
273、步骤1:5-(2-氯-5-甲氧基嘧啶-4-基)-1-甲基-1h-苯并[d]咪唑
274、
275、在混于二恶烷(5ml)和水(5ml)内的2,4-二氯-5-甲氧基嘧啶化合物(600mg,3.37mmol,1.0当量)及(1-甲基-1h-苯并[d]咪唑-5-基)硼酸化合物(652mg,3.71mmol,1.1当量)中,加入pd(dppf)cl2(270mg,0.33mol,0.1当量)及碳酸铯(2.19g,6.74mmol,2.0当量)。混合物在氮气气氛中110℃搅拌4小时。反应混合物的lcms分析表明完全转化至所需产物。混合物过滤后,滤过物溶于乙酸乙酯(8ml)中,并经饱和氯化钠水溶液洗涤(10ml×3)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(二氯甲烷:甲醇=99:1),获得白色固体的5-(2-氯-5-甲氧基嘧啶-4-基)-1-甲基-1h-苯并[d]咪唑化合物(430mg,46.3%)。
276、lc-ms m/z:275.3(m+1)+。
277、步骤2:5-(2-氯-5-甲氧基嘧啶-4-基)-1-甲基-1h-苯并[d]咪唑
278、
279、在混于叔丁醇(5ml)内的5-(2-氯-5-甲氧基嘧啶-4-基)-1-甲基-1h-苯并[d]咪唑化合物(300mg,1.09mmol,1.0当量)、4-(4-甲基哌嗪-1-基)苯胺化合物(300mg,1.58mmol,1.5当量)及碳酸钾(300mg,2.18mmol,2.0当量)中,加入pd2(dba)3(100mg,0.11mmol,0.1当量)及xphos(260mg,0.545mmol,0.2当量)。混合物以氮气脱气3次,并在氮气气氛中100℃搅拌48小时。反应混合物的lcms分析表明反应完成。混合物减压浓缩后,残余物经制备型hplc纯化,获得棕色固体的实施例25(21.4mg,4.5%)。
280、制备型hplc条件:
281、方法a:仪器:岛津lc8ap制备型hplc系统;色谱柱:boston ods(250mm×21.2mm×10μm,);流动相:溶有0.1%甲酸的水/乙腈;梯度:乙腈在1分钟至5分钟时间段为5%,在5分钟至20分钟时间段为5%至25%,在20分钟至25分钟时间段为25%;柱温:25℃;检测波长:214/254nm;流速:20ml/min;
282、lc-ms m/z:430.4(m+1)+;
283、1h nmr(400mhz,cd3od):δ8.52(s,1h),8.46(s,1h),8.30(s,1h),8.24~8.12(m,2h),7.61(d,j=8.4,3h),6.98(d,j=8.4,2h),3.92(s,3h),3.87(s,3h),3.29~3.25(m,4h),3.06~3.02(m,4h),2.65(s,3h)。
284、步骤3:5-(2-氯-5-甲氧基嘧啶-4-基)-1-甲基-1h-苯并[d]咪唑
285、
286、0℃下,在混于dcm(5ml)内的5-(2-氯-5-甲氧基嘧啶-4-基)-1-甲基-1h-苯并[d]咪唑化合物(200mg,0.46mmol,1.0当量)中,缓慢加入三溴化硼(1ml)。混合物以氮气脱气3次,并在氮气气氛中25℃搅拌16小时。反应混合物的lcms分析表明反应完成。混合物经饱和氯化钠水溶液洗涤(8ml×3)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经制备型hplc纯化(a:0.1%甲酸水溶液;b:乙腈),获得黄色固体的实施例26化合物(31.7mg,16.2%)。
287、制备型hplc条件:
288、方法a:仪器:岛津lc8ap制备型hplc系统;色谱柱:boston ods(250mm×21.2mm×10μm,);流动相:溶有0.1%甲酸的水/乙腈;梯度:乙腈在1分钟至5分钟时间段为5%,在5分钟至20分钟时间段为5%至25%,在20分钟至25分钟时间段为25%;柱温:25℃;检测波长:214/254nm;流速:20ml/min;
289、lc-ms m/z:416.5(m+1)+;
290、1h nmr(400mhz,cd3od):δ8.66(s,1h),8.41(s,1h),8.32(d,j=8.4,1h),8.17(s,1h),8.07(s,1h),7.65~7.56(m,3h),6.98(d,j=8.8,2h),3.92(s,3h),3.29(s,4h),3.16(s,4h),2.75(s,3h)。
291、实施例27:
292、5-氯-4-(2-甲基-1h-苯并[d]咪唑-5-基)-n-(4-(4-甲基哌嗪-1-基)苯基)嘧啶-2-胺
293、
294、路线图
295、
296、步骤1:5-溴-2-甲基-1-((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑
297、
298、0℃下,在混于无水dmf(30ml)内的5-溴-2-甲基-1h-苯并[d]咪唑化合物(2g,9.48mmol,1.0当量)中,加入氢化钠(455mg,11.37mol,1.2当量)。混合物在氮气气氛中0℃搅拌0.5小时后,加入semcl(2.05g,12.32mmol,1.3当量),并在氮气气氛中室温搅拌16小时。反应混合物的lcms分析表明完全转化至所需产物。反应以氯化铵水溶液淬灭,残余物溶于二氯甲烷(30ml)中,并经饱和氯化钠水溶液洗涤(40ml×3)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(二氯甲烷:甲醇=0~3%),获得白色固体的5-溴-2-甲基-1-((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑化合物(3.1g,96.8%)。
299、lc-ms m/z:341.2(m+1)+。
300、步骤2:2-甲基-5-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1-((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑
301、
302、在混于二恶烷(20ml)内的5-溴-2-甲基-1-((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑化合物(2.0g,5.85mmol,1.0当量)、b2pin2(1.78g,7.02mmol,1.2当量)及乙酸钾(1.72g,17.54mmol,3.0当量)中,加入pd(dppf)cl2(240mg,0.29mol,0.05当量)。混合物以氮气脱气3次,并在氮气气氛中110℃搅拌10小时。反应混合物的lcms分析表明反应完成。混合物经乙酸乙酯稀释(30ml)、饱和氯化钠水溶液洗涤(5ml)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(二氯甲烷:甲醇=0~3%),获得黄色固体的2-甲基-5-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1-((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑化合物(1.8g,62.5%)。
303、lc-ms m/z:389.5(m+1)+。
304、步骤3:5-(2,5-二氯嘧啶-4-基)-2-甲基-1-((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑
305、
306、在混于thf(30ml)和水(15ml)内的2-甲基-5-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1-((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑化合物(1.6g,5.78mmol,1.0当量)、2,4,5-三氯嘧啶化合物(2.12g,11.56mmol,2.0当量)及碳酸钠(1.83g,17.3mmol,3.0当量)中,加入乙酸钯(130mg,0.56mol,0.1当量)及pph3(303mg,1.16mol,0.2当量)。混合物以氮气脱气3次,并在氮气气氛中80℃搅拌4小时。反应混合物的tlc分析表明反应完成。混合物溶于二氯甲烷(30ml),并经饱和氯化钠水溶液洗涤(40ml×3)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(石油醚:乙酸乙酯=0~30%),获得黄色固体的标题所示化合物(800mg,47.6%)。
307、lc-ms m/z:408.4(m+1)+。
308、步骤4:5-氯-4-(2-甲基-1-((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑-5-基)-n-(4-(4-甲基哌嗪-1-基)苯基)嘧啶-2-胺
309、
310、在混于二恶烷(6ml)内的5-(2,5-二氯嘧啶-4-基)-2-甲基-1-((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑化合物(350mg,0.87mmol,1.0当量)、4-(4-甲基哌嗪-1-基)苯胺化合物(245mg,1.05mmol,1.2当量)及碳酸铯(840mg,3.75mmol,2.61当量)中,加入xantphos-pd-g3(84mg,0.08mol,0.1当量)。混合物以氮气脱气3次,并在氮气气氛中110℃搅拌16小时。反应混合物的lcms分析表明反应完成。混合物经乙酸乙酯稀释(15ml)、饱和氯化钠水溶液洗涤(5ml)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(二氯甲烷:甲醇=0~3%),获得黄色固体的5-氯-4-(2-甲基-1-((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑-5-基)-n-(4-(4-甲基哌嗪-1-基)苯基)嘧啶-2-胺化合物(240mg,49.7%)。
311、lc-ms m/z:564.3(m+1)+。
312、步骤5:5-氯-4-(2-甲基-1h-苯并[d]咪唑-5-基)-n-(4-(4-甲基哌嗪-1-基)苯基)嘧啶-2-胺
313、
314、在混于dcm(4ml)内的5-氯-4-(2-甲基-1-((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑-5-基)-n-(4-(4-甲基哌嗪-1-基)苯基)嘧啶-2-胺化合物(240mg,0.42mmol,1.0当量)中,加入tfa(4ml)。混合物以氮气脱气3次,并在氮气气氛中室温搅拌16小时。反应混合物的lcms分析表明反应完成。混合物经制备型hplc纯化,获得黄色固体的实施例27(45.9mg,24.9%)。
315、制备型hplc条件:
316、方法b:仪器:岛津lc8ap制备型hplc系统;色谱柱:boston ods(250mm×21.2mm×10μm,);流动相:溶于水/乙腈的0.1% tfa;梯度:乙腈在1分钟至5分钟时间段为5%,在5分钟至20分钟时间段为10%至30%,在20分钟至25分钟时间段为30%;柱温:25℃;检测波长:214/254nm;流速:20ml/min;
317、lc-ms m/z:434.4(m+1)+;
318、1h nmr(400mhz,cd3od):δ8.46(s,1h),8.21(s,1h),8.04(d,j=8.8hz,1h),7.83(d,j=8.8,1h),7.61(d,j=8.4hz,2h),6.97(d,j=8.4hz,2h),3.73~3.54(m,2h),3.58(d,j=8.0hz,2h),3.28(d,j=8.4hz,2h),3.04~3.01(m,2h),2.90(s,3h),2.86(s,3h)。
319、实施例28:
320、5-氯-4-(2-甲基-1h-苯并[d]咪唑-5-基)-n-(4-(4-甲基哌嗪-1-基)苯基)嘧啶-2-胺
321、
322、在混于dmf(10ml)内的实施例18化合物(200mg,0.5mmol,1.0当量)中,加入甲硫醇钠(700mg,以20%w/w溶于水,2mmol,4当量)。混合物以氮气脱气3次,并在biotage上140℃搅拌1小时。反应混合物的tlc分析表明形成约50%所需产物。混合物以硅藻土过滤,并经减压浓缩,获得粗产物。粗产物经制备型hplc纯化,获得黄色固体的实施例28(23.3mg,10.9%)。
323、制备型hplc条件:
324、方法b:仪器:岛津lc8ap制备型hplc系统;色谱柱:boston ods(250mm×21.2mm×10μm,);流动相:溶有0.1%盐酸的水/乙腈;梯度:乙腈在1分钟至5分钟时间段为5%,在5分钟至20分钟时间段为10%至30%,在20分钟至25分钟时间段为30%;柱温:25℃;检测波长:214/254nm;流速:20ml/min;
325、lc-ms m/z:433.2(m+1)+;
326、1h nmr(400mhz,cd3od):δ9.55(s,1h),8.62(s,1h),8.37(s,1h),8.14-8.07(m,2h),7.95(d,j=8.0hz,2h),7.76(d,j=8.0hz,2h),4.21(s,3h),4.16-4.13(m,4h),3.73~3.69(m,4h),2.34(s,3h)。
327、实施例29:
328、5-氯-4-(2-甲基-1h-苯并[d]咪唑-5-基)-n-(4-(4-甲基哌嗪-1-基)苯基)嘧啶-2-胺
329、
330、路线图
331、
332、步骤1:2-(2,2-二氟苯并[d][1,3]二氧杂环戊烯-5-基)-4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷
333、
334、在混于二恶烷(20ml)内的5-溴-2,2-二氟苯并[d][1,3]二氧杂环戊烯化合物(2.0g,8.44mmol,1.0当量)、b2pin2(3.0g,11.81mmol,1.4当量)及乙酸钾(1.65g,16.87mmol,2.0当量)中,加入pd(dppf)cl2(695.5mg,0.84mol,0.1当量)。混合物以氮气脱气3次,并在氮气气氛中100℃搅拌3小时。反应混合物的lcms分析表明反应完成。混合物经过滤和浓缩,残余物经乙酸乙酯稀释(20ml)、饱和氯化钠水溶液洗涤(25ml×3)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(石油醚:乙酸乙酯=0~2%),获得灰色固体的2-(2,2-二氟苯并[d][1,3]二氧杂环戊烯-5-基)-4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷(1.65g,66.6%)化合物。
335、lc-ms m/z:285.4(m+1)+。
336、步骤2:2,5-二氯-4-(2,2-二氟苯并[d][1,3]二氧杂环戊烯-5-基)嘧啶
337、
338、在混于thf(12ml)和水(5ml)内的2-(2,2-二氟苯并[d][1,3]二氧杂环戊烯-5-基)-4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷化合物(600mg,2.11mmol,1.0当量)、2,4,5-三氯嘧啶化合物(5.81g,3.17mmol,1.5当量)及碳酸钠(670mg,6.33mmol,3.0当量)中,加入乙酸钯(48mg,0.21mol,0.1当量)及pph3(221mg,0.84mol,0.4当量)。混合物以氮气脱气3次,并在氮气气氛中80℃搅拌4小时。反应混合物的lcms分析表明反应完成。混合物溶于乙酸乙酯(10ml)中,并经饱和氯化钠水溶液洗涤(10ml×3)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(石油醚:乙酸乙酯=0~3%),获得白色固体的2,5-二氯-4-(2,2-二氟苯并[d][1,3]二氧杂环戊烯-5-基)嘧啶化合物(450mg,70.1%)。
339、lc-ms m/z:305.5(m+1)+。
340、步骤3:2,5-二氯-4-(2,2-二氟苯并[d][1,3]二氧杂环戊烯-5-基)嘧啶
341、
342、在混于二恶烷(4ml)内的2,5-二氯-4-(2,2-二氟苯并[d][1,3]二氧杂环戊烯-5-基)嘧啶化合物(200mg,0.65mmol,1.0当量)、4-(4-甲基哌嗪-1-基)苯胺化合物(188mg,0.98mmol,1.5当量)及碳酸铯(640mg,1.96mmol,3当量)中,加入xantphos-pd-g3(62mg,0.06mol,0.1当量)。混合物以氮气脱气3次,并在氮气气氛中110℃搅拌16小时。反应混合物的lcms分析表明反应完成。混合物经乙酸乙酯稀释(15ml)、饱和氯化钠水溶液洗涤(5ml)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经制备型hplc纯化,获得黄色固体的实施例29(31.1mg,10.3%)。
343、制备型hplc条件:
344、方法a:仪器:岛津lc8ap制备型hplc系统;色谱柱:boston ods(250mm×21.2mm×10μm,);流动相:溶有0.1%甲酸的水/乙腈;梯度:乙腈在1分钟至5分钟时间段为5%,在5分钟至20分钟时间段为5%至25%,在20分钟至25分钟时间段为25%;柱温:25℃;检测波长:214/254nm;流速:20ml/min;
345、lc-ms m/z:460.4(m+1)+;
346、1h nmr(400mhz,cd3od):δ8.39(s,1h),7.79-7.62(m,2h),7.53(d,j=8.4hz,2h),7.31(d,j=8.4hz,1h),6.94(d,j=8.4hz,2h),3.14-3.10(m,4h),2.65~2.62(m,4h),2.35(s,3h)。
347、实施例30:
348、4-(苯并[d]噻唑-5-基)-n-(4-吗啉基苯基)嘧啶-2-胺
349、
350、路线图
351、
352、步骤1:5-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)苯并[d]噻唑
353、
354、在混于二恶烷(100ml)内的5-溴苯并[d]噻唑化合物(2.8g,13.1mmol,1.0当量)中,加入双(频哪醇合)二硼(5.0g,19.6mol,1.5当量)、乙酸钾(2.5g,26.2mol,2.0当量)及[1,1'-双(二苯基膦)二茂铁]二氯化钯(540mg,0.65mol,0.05当量)。混合物在氮气气氛中100℃搅拌16小时。反应混合物的lcms分析表明完全转化至所需产物。随后,混合物以水稀释(50ml),并以乙酸乙酯萃取(50ml×2)。有机层经饱和氯化钠水溶液洗涤(50ml)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(石油醚:乙酸乙酯=0~2%),获得白色固体的5-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)苯并[d]噻唑化合物(2.0g,59%)。
355、lc-ms m/z:262.3(m+1)+。
356、步骤2:5-(2-氯嘧啶-4-基)苯并[d]噻唑
357、
358、在混于二恶烷/水(20ml/5ml)内的5-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)苯并[d]噻唑化合物(1.0g,3.8mmol,1.0当量)中,加入2,4-二氯嘧啶化合物(1.7,11.4mmol,3.0当量)、碳酸钠(1.2g,11.4mol,3.0当量)及[1,1'-双(二苯基膦)二茂铁]二氯化钯(155mg,0.19mol,0.05当量)。混合物在氮气气氛中100℃搅拌30分钟。反应混合物的lcms分析表明完全转化至所需产物。反应混合物以水稀释(20ml),并以乙酸乙酯萃取(30ml×2)。有机层经饱和氯化钠水溶液洗涤(30ml×2)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(石油醚:乙酸乙酯=0~2%),获得白色固体的5-(2-氯嘧啶-4-基)苯并[d]噻唑化合物(50mg,5%)。
359、lc-ms m/z:248.4(m+1)+;
360、步骤3:4-(苯并[d]噻唑-5-基)-n-(4-吗啉基苯基)嘧啶-2-胺
361、
362、在混于乙腈(3ml)内的5-(2-氯嘧啶-4-基)苯并[d]噻唑化合物(50mg,0.20mmol,1.0当量)中,加入4-吗啉基苯胺化合物(53mg,0.30mmol,1.5当量)及对甲苯磺酸(46mg,0.24mol,1.2当量)。混合物在氮气气氛中80℃搅拌16小时。反应混合物的lcms分析表明完全转化至所需产物。反应混合物以水稀释(5ml),并以乙酸乙酯萃取(10ml×2)。有机层经饱和氯化钠水溶液洗涤(30ml×2)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经制备型hplc纯化,获得黄色固体的实施例30(31.8mg,32%)。
363、制备型hplc条件:
364、方法a:仪器:岛津lc8ap制备型hplc系统;色谱柱:boston ods(250mm×21.2mm×10μm,);流动相:溶有0.1%甲酸的水/乙腈;梯度:乙腈在1分钟至5分钟时间段为5%,在5分钟至20分钟时间段为5%至25%,在20分钟至25分钟时间段为25%;柱温:25℃;检测波长:214/254nm;流速:20ml/min;
365、lc-ms m/z:390.3(m+1)+;
366、1h nmr(d6-dmso,400mhz):δ9.48(s,1h),9.43(s,1h),8.94(s,1h),8.43(d,j=8.4hz,1h),8.32(d,j=6.0hz,1h),8.24(d,j=8.4hz,1h),7.58(d,j=7.6hz,2h),6.98(d,j=8.8hz,2h),6.62(d,j=6.0hz,1h),3.73~3.70(m,4h),3.08~3.05(m,4h)。
367、实施例31:
368、5-(5-氯-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)-1h-苯并[d]
369、咪唑-2(3h)-酮
370、
371、路线图
372、
373、步骤1:6-(2-氯嘧啶-4-基)-1-甲基-1h-吲唑
374、
375、在混于乙腈/水(20ml/10ml)内的2,4-二氯嘧啶化合物(834mg,5.6mmol,2.0当量)中,加入(1-甲基-1h-吲唑-6-基)硼酸化合物(500mg,2.8mol,1.0当量)、碳酸钠(890mg,8.4mol,3.0当量)及四(三苯基膦)钯(324mg,0.28mol,0.1当量)。混合物在氮气气氛中90℃搅拌2小时。反应混合物的lcms分析表明完全转化至所需产物。随后,混合物以乙酸乙酯萃取(20ml×2),有机层经饱和氯化钠水溶液洗涤(30ml)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(二氯甲烷:甲醇=0~2%),获得白色固体的6-(2-氯嘧啶-4-基)-1-甲基-1h-吲唑化合物(450mg,65%)。
376、lc-ms m/z:245.2(m+1)+。
377、步骤2:5-(5-氯-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)-1h-苯并[d]咪唑-2(3h)-酮
378、
379、在混于乙腈(10ml)内的6-(2-氯嘧啶-4-基)-1-甲基-1h-吲唑化合物(300mg,1.2mmol,1.0当量)中,加入4-吗啉基苯胺化合物(270mg,1.4mmol,1.2当量)及对甲苯磺酸(270mg,1.4mol,1.2当量)。混合物在氮气气氛中80℃搅拌16小时。反应混合物的lcms分析表明完全转化至所需产物。反应混合物以水稀释(30ml),并以乙酸乙酯萃取(30ml×2)。有机层经饱和氯化钠水溶液洗涤(30ml×2)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经制备型hplc纯化,获得黄色固体的实施例31(124.7mg,32%)。
380、制备型hplc条件:
381、方法c:仪器:岛津lc8ap制备型hplc系统;色谱柱:boston ods(250mm×21.2mm×10μm,);流动相:溶有0.1%甲酸的水/乙腈;梯度:乙腈在1分钟至5分钟时间段为5%,在5分钟至20分钟时间段为20%至40%,在20分钟至25分钟时间段为40%;柱温:25℃;检测波长:214/254nm;流速:20ml/min;
382、lc-ms m/z:387.3(m+1)+;
383、1h nmr(d6-dmso,400mhz):δ9.46(s,1h),8.50(d,j=5.2hz,1h),8.41(s,1h),8.09(s,1h),7.89(dd,j=26.4,8.4hz,2h),7.67(d,j=8.8hz,2h),7.46(d,j=4.4hz,1h),6.91(d,j=8.4hz,2h),4.11(s,3h),3.71(t,j=7.6hz,4h),3.02(t,j=7.6hz,4h)。
384、实施例32:
385、4-(苯并[d]恶唑-5-基)-5-氯-n-(4-(4-甲基哌嗪-1-基)苯基)嘧啶-2-胺
386、
387、路线图
388、
389、步骤1:5-(2,5-二氯嘧啶-4-基)苯并[d]恶唑
390、
391、在混于thf(12ml)和水(6ml)内的5-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)苯并[d]恶唑化合物(530mg,2.16mmol,1.0当量)、2,4,5-三氯嘧啶化合物(794mg,4.326mmol,2.0当量)及碳酸钠(688mg,6.48mmol,3当量)中,加入乙酸钯(50mg,0.21mol,0.1当量)及pph3(113mg,0.43mol,0.2当量)。混合物以氮气脱气3次,并在氮气气氛中80℃搅拌4小时。反应混合物的lcms分析表明反应完成。混合物溶于乙酸乙酯(10ml)中,并经饱和氯化钠水溶液洗涤(40ml×3)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(石油醚:乙酸乙酯=0~15%),获得黄色固体的5-(2,5-二氯嘧啶-4-基)苯并[d]恶唑化合物(400mg,69.8%)。
392、lc-ms m/z:266.1(m+1)+;
393、步骤2:4-(苯并[d]恶唑-5-基)-5-氯-n-(4-(4-甲基哌嗪-1-基)苯基)嘧啶-2-胺
394、
395、在混于二恶烷(6ml)内的5-(2,5-二氯嘧啶-4-基)苯并[d]恶唑化合物(200mg,0.75mmol,1.0当量)、4-(4-甲基哌嗪-1-基)苯胺化合物(216mg,1.13mmol,1.5当量)及碳酸铯(738mg,2.26mmol,3当量)中,加入xantphos-pd-g3(72mg,0.07mol,0.1当量)。混合物以氮气脱气3次,并在氮气气氛中110℃搅拌16小时。反应混合物的lcms分析表明反应完成。混合物经乙酸乙酯稀释(15ml)、饱和氯化钠水溶液洗涤(5ml)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经制备型tlc纯化(二氯甲烷:甲醇=10:1),获得黄色固体的实施例32(22.0mg,6.3%)。
396、lc-ms m/z:421.4(m+1)+;
397、1h nmr(400mhz,cd3od):δ8.56(s,1h),8.42(s,1h),8.25(s,1h),7.96(d,j=8.4hz,1h),7.78(d,j=8.8hz,1h),7.58(d,j=8.4hz,2h),6.95(d,j=8.8hz,2h),3.20-3.13(m,4h),2.85~2.38(m,4h),2.51(s,3h)。
398、实施例33:
399、5-(5-氯-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)-1h-苯并[d]咪唑-2(3h)-酮
400、
401、路线图
402、
403、步骤1:6-溴-1-(甲氧基甲基)-1h-吲唑
404、
405、0℃下,在混于n,n-二甲基甲酰胺(40ml)内的6-溴-1h-吲唑化合物(2.0g,10.1mmol,1.0当量)中,加入氢化钠(292mg,12.1mmol,1.2当量)。混合物在0℃下搅拌0.5小时后,加入氯甲基甲醚(980mg,12.1mmol,1.2当量),并室温搅拌2小时。反应混合物的lcms分析表明完全转化至所需产物。反应混合物以水稀释(30ml),并以乙酸乙酯萃取(30ml×2)。有机层经饱和氯化钠水溶液洗涤(50ml)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(石油醚:乙酸乙酯=0~10%),获得白色固体的6-溴-1-(甲氧基甲基)-1h-吲唑化合物(1.0g,42%)。
406、lc-ms m/z:241.2(m+1)+。
407、步骤2:1-(甲氧基甲基)-6-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1h-吲唑
408、
409、在混于二甲亚砜(30ml)内的6-溴-1-(甲氧基甲基)-1h-吲唑化合物(1.0g,4.2mmol,1.0当量)中,加入b2pin2(1.58g,6.2mmol,1.5当量)、乙酸钾(816mg,8.3mol,2.0当量)及[1,1'-双(二苯基膦)二茂铁]二氯化钯(170mg,0.2mol,0.05当量)。混合物在氮气气氛中80℃搅拌2小时。反应混合物的lcms分析表明完全转化至所需产物。反应混合物以水稀释(20ml),并以乙酸乙酯萃取(30ml×2)。有机层经饱和氯化钠水溶液洗涤(30ml×2)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(二氯甲烷:甲醇=0~2%),获得棕色固体的1-(甲氧基甲基)-6-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1h-吲唑化合物(1.0g,84%)。
410、lc-ms m/z:289.7(m+1)+。
411、步骤3:6-(2,5-二氯嘧啶-4-基)-1-(甲氧基甲基)-1h-吲唑
412、
413、在混于四氢呋喃/水(6ml/2ml)内的1-(甲氧基甲基)-6-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1h-吲唑化合物(200mg,0.7mmol,1.0当量)中,加入2,4,5-三氯嘧啶化合物(127mg,0.7mol,1.0当量)、碳酸钠(220mg,2.1mol,3.0当量)、乙酸钯(16mg,0.07mol,0.1当量)及三苯基磷(72.7mg,0.3mol,0.4当量)。混合物在氮气气氛中回流搅拌3小时。反应混合物的lcms分析表明完全转化至所需产物。随后,混合物以水稀释(10ml),并以乙酸乙酯萃取(10ml×2)。有机层经饱和氯化钠水溶液洗涤(10ml)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(二氯甲烷:甲醇=0~2%),获得棕色固体的6-(2,5-二氯嘧啶-4-基)-1-(甲氧基甲基)-1h-吲唑化合物(150mg,70%)。
414、lc-ms m/z:309.5(m+1)+。
415、步骤4:5-氯-4-(1-(甲氧基甲基)-1h-吲唑-6-基)-n-(4-(4-甲基哌嗪-1-基)苯基)嘧啶-2-胺
416、
417、在混于二恶烷(10ml)内的6-(2,5-二氯嘧啶-4-基)-1-(甲氧基甲基)-1h-吲唑化合物(400mg,1.3mmol,1.0当量)中,加入4-(4-甲基哌嗪-1-基)苯胺化合物(247mg,1.3mmol,1.0当量)、碳酸铯(1.27g,3.9mmol,3.0当量)及xphos-pd-g3(61mg,0.064mmol,0.05当量)。混合物在氮气气氛中中110℃搅拌16小时。反应混合物的lcms分析表明完全转化至所需产物。反应混合物以水稀释(5ml),并以乙酸乙酯萃取(10ml×2)。有机层经饱和氯化钠水溶液洗涤(10ml×2)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(二氯甲烷:甲醇=0~3%),获得黄色固体的5-氯-4-(1-(甲氧基甲基)-1h-吲唑-6-基)-n-(4-(4-甲基哌嗪-1-基)苯基)嘧啶-2-胺化合物(280mg,47%)。
418、lc-ms m/z:464.2(m+1)+。
419、步骤5:5-氯-4-(1h-吲唑-6-基)-n-(4-(4-甲基哌嗪-1-基)苯基)嘧啶-2-胺
420、
421、在混于异丙醇(ipa)(5ml)内的5-氯-4-(1-(甲氧基甲基)-1h-吲唑-6-基)-n-(4-(4-甲基哌嗪-1-基)苯基)嘧啶-2-胺化合物(260mg,0.56mmol,1.0当量)中,加入三氟乙酸(127mg,1.11mmol,2.0当量)。混合物在90℃下搅拌16小时。反应混合物的lcms分析表明完全转化至所需产物。反应混合物经减压浓缩,获得粗产物。粗产物经制备型hplc纯化,获得橙色固体的实施例33(25mg,11%)。
422、方法b:仪器:岛津lc8ap制备型hplc系统;色谱柱:boston ods(250mm×21.2mm×10μm,);流动相:溶有0.1%甲酸的水/乙腈;梯度:乙腈在1分钟至5分钟时间段为5%,在5分钟至20分钟时间段为10%至30%,在20分钟至25分钟时间段为30%;柱温:25℃;检测波长:214/254nm;流速:20ml/min;
423、lc-ms m/z:420.4(m+1)+;
424、1h nmr(400mhz,cd3od)δ8.44(s,1h),8.13(s,1h),8.03(s,1h),7.87(d,j=8.4hz,1h),7.62(m,4h),6.98(d,j=8.8hz,2h),3.74(d,j=10.0hz,2h),3.58(d,j=11.2hz,2h),3.25(d,j=10.8hz,2h),3.02~2.95(m,5h)。
425、实施例34:
426、5-(5-氯-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)-1h-苯并[d]
427、咪唑-2(3h)-酮
428、
429、路线图
430、
431、步骤1:3-双((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑-2(3h)-酮
432、
433、0℃下,在混于dmf(50ml)内的5-溴-1h-苯并[d]咪唑-2(3h)-酮化合物(2.13g,10.0mmol,1.0当量)中,加入氢化钠(880mg,以60%散于矿物油中,22.2mmol,2.2当量)。搅拌0.5小时后,缓慢加入semcl(3.67g,2.2mol,2.2当量)。混合物在氮气气氛中室温搅拌16小时。反应混合物的tlc分析表明反应完成。混合物以饱和氯化铵(10ml)淬灭,并经乙酸乙酯稀释(400ml)、卤水洗涤(200ml×3)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(石油醚:乙酸乙酯=0~20%),获得无色油状物的5-溴-1,3-双((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑-2(3h)-酮化合物(2.1g,44.5%)。
434、lc-ms m/z:473.3(m+1)+。
435、步骤2:5-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1,3-双((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑-2(3h)-酮
436、
437、在混于二恶烷(30ml)内的5-溴-1,3-双((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑-2(3h)-酮化合物(2.1g,4.45mmol,1.0当量)、b2pin2(1.36g,5.33mmol,1.3当量)及乙酸钾(873mg,8.90mmol,2.0当量)中,加入pd(dppf)cl2(100mg,0.1mol,0.2当量)。混合物以氮气脱气3次,并在氮气气氛中100℃搅拌3小时。反应混合物的lcms分析表明反应完成。混合物进行过滤和浓缩,残余物经乙酸乙酯稀释(60ml)、卤水洗涤(50ml)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(石油醚:乙酸乙酯=0~20%),获得米白色固体的5-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1,3-双((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑-2(3h)-酮化合物(2.1g,90.7%)。
438、lc-ms m/z:521.3(m+1)+。
439、步骤3:5-(2,5-二氯嘧啶-4-基)-1,3-双((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑-2(3h)-酮
440、
441、在混于thf(6ml)和水(2ml)内的5-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1,3-双((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑-2(3h)-酮化合物(260mg,0.5mmol,1.0当量)、2,4,5-三氯嘧啶化合物(110mg,0.6mmol,1.2当量)及碳酸钠(160mg,1.5mmol,3.0当量)中,加入乙酸钯(11.2mg,0.05mol,0.1当量)及pph3(26mg,0.1mol,0.2当量)。混合物以氮气脱气3次,并在氮气气氛中80℃搅拌16小时。反应混合物的lcms分析表明反应完成。混合物溶于乙酸乙酯(30ml)中,并经卤水洗涤(20ml×3)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(二氯甲烷:甲醇=0~4%),获得淡黄色固体的5-(2,5-二氯嘧啶-4-基)-1,3-双((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑-2(3h)-酮化合物(128mg,70.1%)。
442、lc-ms m/z:541.5(m+1)+。
443、步骤4:5-(5-氯-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)-1,3-双((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑-2(3h)-酮
444、
445、在混于二恶烷(3ml)中的5-(2,5-二氯嘧啶-4-基)-1,3-双((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑-2(3h)-酮化合物(128mg,0.24mmol,1.0当量)、4-(4-甲基哌嗪-1-基)苯胺化合物(46mg,0.24mmol,1.0当量)及碳酸铯(235mg,0.72mmol,3当量)中,加入xantphos-pd-g3(100mg,0.1mmol,0.1当量)。混合物以氮气脱气3次,并在氮气气氛中110℃搅拌16小时。反应混合物的lcms分析表明反应完成。混合物以硅藻土过滤,并经减压浓缩,获得黑色固体的5-(5-氯-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)-1,3-双((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑-2(3h)-酮(粗产物,500mg,100%)粗产化合物。该粗产物直接用于下一步骤,无需进一步纯化。
446、lc-ms m/z:696.4(m+1)+。
447、步骤5:5-(5-氯-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)-1h-苯并[d]咪唑-2(3h)-酮
448、
449、将溶于tfa(3ml)和二氯甲烷(3ml)中的5-(5-氯-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)-1,3-双((2-(三甲基硅烷基)乙氧基)甲基)-1h-苯并[d]咪唑-2(3h)-酮化合物(粗产物,500mg,0.24mmol,1.0当量)室温搅拌1小时。反应混合物的tlc分析表明反应完成。混合物经减压浓缩后,残余物悬于氨水(10ml)中,并室温搅拌1小时。反应混合物的lcms分析表明反应完成。混合物经减压浓缩,获得粗产物。粗产物经制备型hplc纯化,获得黄色固体的实施例34(4.7mg,4.5%)。
450、制备型hplc条件:
451、方法b:仪器:岛津lc8ap制备型hplc系统;色谱柱:boston ods(250mm×21.2mm×10μm,);流动相:溶有0.1%甲酸的水/乙腈;梯度:乙腈在1分钟至5分钟时间段为5%,在5分钟至20分钟时间段为10%至30%,在20分钟至25分钟时间段为30%;柱温:25℃;检测波长:214/254nm;流速:20ml/min;
452、lc-ms m/z:436.4(m+1)+;
453、1h nmr(400mhz,d6-dmso):δ10.88(s,1h),10.80(s,1h),9.58(s,1h),8.45(s,1h),8.21(s,1h),7.53(d,j=8.0hz,2h),7.48(d,j=8.0hz,1h),7.40(s,1h),7.02(d,j=8.0hz,1h),6.84(d,j=8.4hz,2h),3.04~3.00(m,4h),2.43~2.38(m,4h),2.17(s,3h)。
454、实施例35:
455、4-(1-甲基-1h-苯并[d]咪唑-5-基)-n-(4-吗啉基苯基)嘧啶-2-胺
456、
457、路线图
458、
459、步骤1:1-甲基-5-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1h-苯并[d]咪唑
460、
461、氮气气氛中,在混于二恶烷(40ml)内的5-溴-1-甲基-1h-苯并[d]咪唑化合物(2.0g,9.5mmol,1.0当量)及b2pin2(3.6g,14.2mmol,1.0当量)中,加入乙酸钾(1.86g,19.0mmol,2.0当量)及pd(dppf)cl2(781mg,0.95mmol,0.1当量)。混合物在氮气气氛中110℃搅拌16小时。反应混合物的hplc和lcms分析表明完全转化至所需产物。混合物以水稀释(40ml),并以乙酸乙酯萃取(30ml×2)。有机层经无水硫酸钠干燥、过滤及减压浓缩。残余物经硅胶柱层析纯化(二氯甲烷:甲醇=0~3%),获得黄色固体的1-甲基-5-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1h-苯并[d]咪唑化合物(2.0g,82%)。
462、lc-ms m/z:259.4(m+1)+。
463、步骤2:5-(2-氯嘧啶-4-基)-1-甲基-1h-苯并[d]咪唑
464、
465、氮气气氛中,在混于乙腈(20ml)内的1-甲基-5-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1h-苯并[d]咪唑化合物(800mg,3.1mmol,1.0当量)及2,4-二氯嘧啶化合物(924mg,6.2mmol,2.0当量)中,加入碳酸钠溶液(20ml,0.4mol/l)及pd(pph3)4(358mg,0.31mmol,0.1当量)。混合物在氮气气氛中90℃搅拌4小时。反应混合物的lcms分析表明完全转化至所需产物。混合物以水稀释(20ml),并以乙酸乙酯萃取(20ml×2)。有机层经无水硫酸钠干燥、过滤及减压浓缩。残余物经硅胶柱层析纯化(二氯甲烷:甲醇=0~3%),获得黄色固体的5-(2-氯嘧啶-4-基)-1-甲基-1h-苯并[d]咪唑化合物(560mg,74%)。
466、lc-ms m/z:245.3(m+1)+。
467、步骤3:4-(1-甲基-1h-苯并[d]咪唑-5-基)-n-(4-吗啉基苯基)嘧啶-2-胺
468、
469、氮气气氛中,在混于异丙醇(10ml)内的5-(2-氯嘧啶-4-基)-1-甲基-1h-苯并[d]咪唑化合物(500mg,2.0mmol,1.0当量)及4-吗啉基苯胺化合物(726mg,4.0mmol,2.0当量)中,加入tfa(230mg,20mmol,10当量)。混合物在氮气气氛中100℃搅拌16小时。反应混合物的tlc分析表明完全转化至所需产物。混合物以水稀释(40ml),并以乙酸乙酯萃取(30ml×2)。有机层经无水硫酸钠干燥、过滤及减压浓缩。残余物经制备型hplc纯化,获得灰色固体的实施例35(102mg,13%)。
470、制备型hplc条件:
471、方法b:仪器:岛津lc8ap制备型hplc系统;色谱柱:boston ods(250mm×21.2mm×10μm,);流动相:溶有0.1%甲酸的水/乙腈;梯度:乙腈在1分钟至5分钟时间段为5%,在5分钟至20分钟时间段为10%至30%,在20分钟至25分钟时间段为30%;柱温:25℃;检测波长:214/254nm;流速:20ml/min;
472、lc-ms m/z:387.2(m+1)+。
473、实施例36:
474、5-氯-n-(4-吗啉基苯基)-4-(1-(2,2,2-三氟乙基)-1h-苯并[d]咪唑-5-基)嘧啶-2-胺与5-氯-n-(4-吗啉基苯基)-4-(1-(2,2,2-三氟乙基)-1h-苯并[d]咪唑-6-基)嘧啶-2-胺
475、
476、路线图
477、
478、步骤1:5-溴-1-(2,2,2-三氟乙基)-1h-苯并[d]咪唑与6-溴-1-(2,2,2-三氟乙基)-1h-苯并[d]咪唑
479、
480、将悬于dmf(30ml)中的5-溴-1h-苯并[d]咪唑(2.0g,10.2mmol,1.0当量)、三氟甲磺酸2,2,2-三氟乙酯(2.6g,11.3mmol,1.1当量)及碳酸铯(6.52g,20.4mmol,2当量)在90℃下搅拌16小时。反应混合物的lcms分析表明完全转化至所需产物。混合物经过滤、乙酸乙酯稀释(150ml)及卤水萃取(3×50ml)。合并有机层经无水硫酸钠干燥、过滤及减压浓缩,获得黄色固体的标题所示化合物(2.7g,100%)。该化合物直接用于下一步骤,无需进一步纯化。
481、lc-ms m/z:279.1(m+1)+。
482、步骤2:5-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1-(2,2,2-三氟乙基)-1h-苯并[d]咪唑与6-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1-(2,2,2-三氟乙基)-1h-苯并[d]咪唑
483、
484、在溶于dmso(30ml)内的5-溴-1-(2,2,2-三氟乙基)-1h-苯并[d]咪唑(2.7g,10mmol,1.0当量)、乙酸钾(3.0g,30mmol,3.0当量)及b2pin2(2.8g,11mmol,1.1当量)中,加入pd(dppf)cl2(946mg,0.1mmol,0.1当量)。混合物在80℃下搅拌8小时。反应混合物的lcms分析表明反应完成。混合物经过滤、乙酸乙酯稀释(150ml)及卤水萃取(3×50ml)。合并有机层经无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(二氯甲烷:甲醇=0~1%),获得黄色固体的5-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1-(2,2,2-三氟乙基)-1h-苯并[d]咪唑(2.8g,88.9%)。
485、lc-ms m/z:327.3(m+1)+。
486、步骤3:5-(2,5-二氯嘧啶-4-基)-1-(2,2,2-三氟乙基)-1h-苯并[d]咪唑与5-(2,5-二氯嘧啶-4-基)-1-(2,2,2-三氟乙基)-1h-苯并[d]咪唑
487、
488、在溶于thf/水(20ml,3:1)内的5-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1-(2,2,2-三氟乙基)-1h-苯并[d]咪唑(0.9g,2.9mmol,1.0当量)、2,4,5-三氯嘧啶(0.66g,3.6mmol,1.2当量)、碳酸钠(660mg,6.2mmol,2.1当量)及pph3(100mg,0.38mmol,0.11当量)中,加入乙酸钯(45mg,0.2mmol,0.06当量)。混合物回流搅拌3小时。反应混合物的lcms分析表明反应完成。混合物经过滤、乙酸乙酯稀释(150ml)及卤水萃取(3×50ml),合并有机层经无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(二氯甲烷:甲醇=0~3%),获得黄色固体的5-(2,5-二氯嘧啶-4-基)-1-(2,2,2-三氟乙基)-1h-苯并[d]咪唑(0.75g,88.9%)。
489、lc-ms m/z:347.2(m+1)+。
490、步骤4:5-氯-n-(4-吗啉基苯基)-4-(1-(2,2,2-三氟乙基)-1h-苯并[d]咪唑-5-基)嘧啶-2-胺与5-氯-n-(4-吗啉基苯基)-4-(1-(2,2,2-三氟乙基)-1h-苯并[d]咪唑-6-基)嘧啶-2-胺
491、
492、在溶于二恶烷(5ml)内的5-(2,5-二氯嘧啶-4-基)-1-(2,2,2-三氟乙基)-1h-苯并[d]咪唑(250mg,0.87mmol,1.0当量)、4-吗啉基苯胺
493、(170mg,0.96mmol,1.1当量)及碳酸铯(851mg,2.6mmol,3当量)中,加入xantphos-pd-g3(946mg,0.1mmol,0.1当量)。混合物在100℃下搅拌8小时。反应混合物的lcms分析表明反应完成。混合物过滤后,滤过物经减压浓缩,获得粗产物。粗产物经制备型hplc纯化,获得棕色固体的实施例36(42.3mg,15.3%)。
494、制备型hplc条件:
495、仪器:岛津lc8ap制备型hplc系统;色谱柱:boston ods(250mm×21.2mm×10μm,);流动相:溶有0.1%甲酸的水/乙腈;梯度:乙腈在1分钟至5分钟时间段为5%,在5分钟至20分钟时间段为5%至25%,在20分钟至25分钟时间段为25%;柱温:25℃;检测波长:214/254nm;流速:20ml/min;
496、lc-ms m/z:489.2(m+1)+;
497、1h nmr(400mhz,cd3od):δ8.41(d,j=3.2hz,1h),8.37(d,j=6.4hz,1h),8.26~8.23(m,1h),7.91(m,1h),7.78(dd,j=16.8,8.4hz,1h),7.59(dd,j=8.8hz,2.4hz,2h),6.93(dd,j=8.8,2.8hz,2h),5.24(q,j=8.8hz,2h),3.81(m,4h),3.07(m,4h)。
498、实施例37:
499、2-(5-(5-氯-2-((4-吗啉基苯基)氨基)嘧啶-4-基)-1h-苯并[d]咪唑-1-基)乙醇
500、
501、路线图
502、
503、步骤1:2-(5-溴-1h-苯并[d]咪唑-1-基)乙醇与2-(6-溴-1h-苯并[d]咪唑-1-基)乙-1-醇
504、
505、在5-溴-1h-苯并[d]咪唑(5.0g,25.4mmol,1.0当量)混合物中,加入1,3-二氧杂环戊烯-2-酮(4.47g,50.76mmol,2.0当量)。混合物以氮气脱气3次,且在氮气气氛中120℃搅拌5小时。反应混合物的lcms分析表明完全转化至所需产物。混合物经减压浓缩,去除溶剂。残余物溶于二氯甲烷(60ml)中,并经饱和氯化钠水溶液洗涤(50ml×2)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(二氯甲烷:甲醇=0~3%),获得淡黄色固体的2-(5-溴-1h-苯并[d]咪唑-1-基)乙醇(3.5g,70%)。
506、lc-ms m/z:241.1(m+1)+。
507、步骤2:2-(5-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1h-苯并[d]咪唑-1-基)乙醇与2-(6-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1h-苯并[d]咪唑-1-基)乙-1-醇
508、
509、在混于dmso(20ml)内的2-(5-溴-1h-苯并[d]咪唑-1-基)乙醇(3.5g,14.5mmol,1.0当量)、b2pin2(3.89g,15.3mmol,1.05当量)及乙酸钾(4.26g,43.5mmol,3.0当量)中,加入pd(dppf)cl2(597mg,0.725mol,0.05当量)。混合物以氮气脱气3次,并在氮气气氛中80℃搅拌4小时。反应混合物的lcms分析表明反应完成。混合物经乙酸乙酯稀释(15ml)、饱和氯化钠水溶液洗涤(5ml)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(二氯甲烷:甲醇=0~3%),获得浅白色固体的2-(5-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1h-苯并[d]咪唑-1-基)乙醇(800mg,57%)。
510、lc-ms m/z:289.3(m+1)+。
511、步骤3:2-(5-(2,5-二氯嘧啶-4-基)-1h-苯并[d]咪唑-1-基)乙醇与2-(6-(2,5-二氯嘧啶-4-基)-1h-苯并[d]咪唑-1-基)乙-1-醇
512、
513、在混于无水thf(15ml)内的2-(5-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1h-苯并[d]咪唑-1-基)乙醇(2.0g,6.94mmol,1.0当量)、2,4,5-三氯嘧啶(1.4g,7.64mmol,1.1当量)、碳酸钠(2.21g,20.82mmol,3.0当量)及pph3(727mg,2.776mmol,0.4当量)中,加入乙酸钯(311mg,1.39mol,0.2当量)。混合物以氮气脱气3次,并在氮气气氛中回流搅拌4小时。反应混合物的lcms分析表明完全转化至所需产物。混合物经减压浓缩,去除溶剂。残余物溶于二氯甲烷(30ml)中,并经饱和氯化钠水溶液洗涤(50ml×2)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(二氯甲烷:甲醇=0~10%),获得淡黄色固体的2-(5-(2,5-二氯嘧啶-4-基)-1h-苯并[d]咪唑-1-基)乙醇(400mg,43%)。
514、lc-ms m/z:309.1(m+1)+。
515、步骤4:2-(5-(5-氯-2-((4-吗啉基苯基)氨基)嘧啶-4-基)-1h-苯并[d]咪唑-1-基)乙醇
516、
517、在混于乙腈(10ml)内的2-(5-(2,5-二氯嘧啶-4-基)-1h-苯并[d]咪唑-1-基)乙醇(300mg,0.97mmol,1.0当量)、4-吗啉基苯胺(260mg,1.46mmol,1.5当量)及对甲苯磺酸(285mg,1.46mmol,1.5当量)中。混合物以氮气脱气3次,并在氮气气氛中90℃搅拌16小时。反应混合物的lcms分析表明反应完成。混合物经乙酸乙酯稀释(15ml)、饱和氯化钠水溶液洗涤(5ml)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经制备型hplc纯化获得棕色固体的
518、实施例37(90.1mg,18%)。
519、制备型hplc条件:
520、仪器:岛津lc8ap制备型hplc系统;色谱柱:boston ods(250mm×21.2mm×10μm,);流动相:溶有0.1%甲酸的水/乙腈;梯度:乙腈在1分钟至5分钟时间段为5%,在5分钟至20分钟时间段为10%至30%,在20分钟至25分钟时间段为30%;柱温:25℃;检测波长:214/254nm;流速:20ml/min;
521、lc-ms m/z:266.3(1/2m+1)+;
522、1h nmr(400mhz,d6-dmso):δ9.60(s,1h),9.64(s,0.1h),8.52(s,1h),8.54(s,0.1h),8.29(s,1h),8.27(s,0.1h),8.11(s,1h),8.07(d,j=12.8,0.1h),7.70(dt,j=29.6,8.4,1h),7.59(d,j=8.8,2h),7.44(d,j=7.6,1h),7.08(d,j=7.2,0.1h),6.87(d,j=8.4,2h),5.05~5.01(m,1h),4.35~4.32(m,2h),3.76-3.70(m,j=4.8,6h),2.99~2.95(m,4h)。
523、实施例38:
524、4-(1-甲基-1h-苯并[d]咪唑-5-基)-n-(4-吗啉基苯基)-5-乙烯基嘧啶-2-胺
525、
526、路线图
527、
528、步骤1:5-氯-4-(1-甲基-1h-苯并[d]咪唑-5-基)-n-(4-吗啉基苯基)嘧啶-2-胺
529、
530、在混于dmf内的5-(2,5-二氯嘧啶-4-基)-1-甲基-1h-苯并[d]咪唑(1.0g,3.59mmol,1.0当量)、4-吗啉基苯胺(640mg,3.59mmol,1.0当量)及磷酸钾(1.52g,7.16mmol,3.0当量)中,加入乙酸钯(80mg,0.357mmol,0.1当量)及xantphos(415mg,0.70mmol,0.2当量)。混合物以氮气脱气3次,并在氮气气氛中120℃搅拌0.5小时。反应混合物的lcms分析表明完全转化至所需产物。混合物经减压浓缩,去除溶剂。残余物溶于中乙酸乙酯(60ml),并经饱和氯化钠水溶液洗涤(50ml×2)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(二氯甲烷:甲醇=0~1%),获得淡黄色固体的5-氯-4-(1-甲基-1h-苯并[d]咪唑-5-基)-n-(4-吗啉基苯基)嘧啶-2-胺(420mg,28%)。
531、lc-ms m/z:421.3(m+1)+。
532、步骤2:4-(1-甲基-1h-苯并[d]咪唑-5-基)-n-(4-吗啉基苯基)-5-乙烯基嘧啶-2-胺
533、
534、在混于thf内的5-氯-4-(1-甲基-1h-苯并[d]咪唑-5-基)-n-(4-吗啉基苯基)嘧啶-2-胺(100mg,0.238mmol,1.0当量)、乙烯三氟硼酸钾(38mg,0.280mmol,1.2当量)及磷酸钾(150mg,0.707mmol,3.0当量)中,加入氯[(4,5-双(二苯基膦基)-9,9-二甲基氧杂蒽)-2-(2'-氨基-1,1'-联苯基)]钯(ii)(xantphos-pd-g2)(20mg,0.026mmol,0.1当量)。混合物以氮气脱气3次,并在氮气气氛中80℃搅拌0.5小时。反应混合物的lcms分析表明完全转化至所需产物。反应混合物经过滤及减压浓缩,获得粗产物。粗产物经制备型hplc纯化,获得淡黄色固体的实施例38(29.8mg,30%)。
535、制备型hplc条件:
536、岛津lc8ap制备型hplc系统;色谱柱:boston ods(250mm×21.2mm×10μm,);流动相:溶有0.1%甲酸的水/乙腈;梯度:乙腈在1分钟至5分钟时间段为5%,在5分钟至20分钟时间段为5%至25%,在20分钟至25分钟时间段为25%;柱温:25℃;检测波长:214/254nm;流速:20ml/min;
537、lc-ms m/z:413.3(m+1)+;
538、1h nmr(400mhz,cd3od):δ9.55(s,1h),8.68(s,1h),8.32(s,1h),7.85(s,1h),7.72~7.49(m,4h),6.86(d,j=8.8hz,2h),6.65~6.53(m,1h),5.74(d,j=17.6hz,1h),5.16(d,j=11.2hz,1h),3.87(s,3h),3.69~3.65(m,4h),2.99~2.96(m,4h)。
539、实施例39:
540、5-甲基-4-(1-甲基-1h-苯并[d]咪唑-5-基)-n-(4-(4-甲基哌嗪-1-基)苯基)嘧啶-2-胺
541、
542、路线图
543、
544、步骤1:5-(2-氯-5-甲基嘧啶-4-基)-1-甲基-1h-苯并[d]咪唑
545、
546、在混于二恶烷(10ml)和水(10ml)内的2,4-二氯-5-甲基嘧啶(400mg,2.36mmol,1.0当量)、(1-甲基-1h-苯并[d]咪唑-5-基)硼酸(590mg,2.13mmol,0.9当量)及碳酸铯(1.5g,4.62mmol,2.0当量)中,加入pd(dppf)cl2(100mg,0.1mol,0.05当量)。混合物以氮气脱气3次,并在氮气气氛中110℃搅拌6小时。反应混合物的lcms分析表明反应完成。混合物溶于中乙酸乙酯(40ml),并经卤水洗涤(40ml×3)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(二氯甲烷:甲醇=0~3%),获得黄色固体的5-(2-氯-5-甲基嘧啶-4-基)-1-甲基-1h-苯并[d]咪唑(425mg,77.9%)。
547、lc-ms m/z:258.5(m+1)+。
548、步骤2:5-甲基-4-(1-甲基-1h-苯并[d]咪唑-5-基)-n-(4-(4-甲基哌嗪-1-基)苯基)嘧啶-2-胺
549、
550、在5-(2-氯-5-甲基嘧啶-4-基)-1-甲基-1h-苯并[d]咪唑(400mg,1.6mmol,1.0当量)、4-(4-甲基哌嗪-1-基)苯胺(330mg,1.7mmol,1.1当量)及碳酸铯(1.47mg,4.6mmol,3.0当量)的混合物中,加入xantphos-pd-g3(100mg,0.1mmol,0.1当量)。混合物以氮气脱气3次,并在biotage上110℃搅拌0.5小时。反应混合物的tlc分析表明反应完成。混合物以硅藻土过滤,并经减压浓缩,获得粗产物。粗产物经制备型hplc纯化,获得棕色固体的实施例39(33.5mg,5.2%)。
551、制备型hplc条件:
552、仪器:岛津lc8ap制备型hplc系统;色谱柱:boston ods(250mm×21.2mm×10μm,);流动相:溶于水/乙腈的0.1% tfa;梯度:乙腈在1分钟至5分钟时间段为5%,在5分钟至20分钟时间段为5%至30%,在20分钟至25分钟时间段为30%;柱温:25℃;检测波长:214/254nm;流速:20ml/min;
553、lc-ms m/z:414.5(m+1)+;
554、1h nmr(400mhz,cd3od):δ9.38(s,1h),8.34(s,1h),8.13(s,1h),8.02(d,j=8.4hz,3h),7.96(d,j=8.4hz,1h),7.58-7.55(m,2h),7.03-6.99(m,2h),4.17(s,3h),3.76-3.74(m,2h),3.59~3.56(m,2h),3.26-3.22(m,2h),2.99~2.96(m,2h),2.95(s,3h),2.28(s,3h)。
555、实施例40:
556、5-氯-4-(1-甲基-1h-苯并[d]咪唑-5-基)-n-(4-吗啉基苯基)吡啶-2-胺
557、
558、路线图
559、
560、步骤1:5-(2,5-二氯吡啶-4-基)-1-甲基-1h-苯并[d]咪唑
561、
562、在混于二恶烷(12ml)和水(3ml)内的4-溴-2,5-二氯吡啶(500mg,2.2mmol,1.0当量)、1-甲基-5-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1h-苯并[d]咪唑(850mg,3.3mmol,1.5当量)、碳酸钠(640mg,6.6mmol,3.0当量)及pd(dppf)cl2(100mg,0.11mmol,0.05当量)中。混合物以氮气脱气3次,并在氮气气氛中100℃搅拌16小时。反应混合物的lcms分析表明完全转化至所需产物。混合物经过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(二氯甲烷:甲醇=0~10%),获得黄色固体的5-(2,5-二氯吡啶-4-基)-1-甲基-1h-苯并[d]咪唑(450mg,73.2%)。
563、lc-ms m/z:278.1(m+1)+。
564、步骤2:5-氯-4-(1-甲基-1h-苯并[d]咪唑-5-基)-n-(4-吗啉基苯基)吡啶-2-胺
565、
566、在混于二恶烷(3ml)内的5-(2,5-二氯吡啶-4-基)-1-甲基-1h-苯并[d]咪唑(300mg,1.08mmol,1.0当量)、4-吗啉基苯胺(211mg,1.19mmol,1.1当量)及碳酸铯(1.05g,3.24mmol,3.0当量)中,加入xantphos-pd-g3(102.5mg,0.11mmol,1.1当量)。混合物以氮气脱气3次,并在氮气气氛中110℃搅拌10小时。反应混合物的lcms分析表明反应完成。混合物经乙酸乙酯稀释(15ml)、饱和氯化钠水溶液洗涤(5ml)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经制备型hplc纯化,获得棕色固体的实施例40化合物(28.0mg,6.0%)。
567、制备型hplc条件:
568、仪器:岛津lc8ap制备型hplc系统;色谱柱:boston ods(250mm×21.2mm×10μm,);流动相:溶有0.1%甲酸的水/乙腈;梯度:乙腈在1分钟至5分钟时间段为5%,在5分钟至20分钟时间段为10%至30%,在20分钟至25分钟时间段为30%;柱温:25℃;检测波长:214/254nm;流速:20ml/min;
569、lc-ms m/z:420.2(m+1)+;
570、1h nmr(400mhz,cdcl3):δ8.20(s,1h),7.92(s,1h),7.82(s,1h),7.42(s,2h),7.20(d,j=8.4hz,2h),6.88(d,j=8.4hz,2h),6.72(s,1h),6.52(s,1h),3.88(s,1h),3.87~3.82(m,8h),3.12~3.08(s,4h)。
571、实施例41:
572、4-(1-甲基-1h-苯并[d]咪唑-5-基)-n2-(4-(4-甲基哌嗪-1-基)苯基)嘧啶-2,5-二胺
573、
574、路线图
575、
576、步骤1:2-氯-4-(1-甲基-1h-苯并[d]咪唑-5-基)嘧啶-5-胺
577、
578、在混于二恶烷/水(9ml/3ml)内的2,4-二氯嘧啶-5-胺(1.0g,6.1mmol,1.00当量)及(1-甲基-1h-苯并[d]咪唑-5-基)硼酸(1.4g,5.0mmol,0.8当量)中,加入碳酸钠(1.6g,6.1mmol,2.50当量)及pd(dppf)cl2(210.0mg,0.25mmol,0.04当量)。混合物以氮气脱气3次,并在氮气气氛中110℃搅拌16小时。反应混合物的lcms分析表明完全转化至所需产物。混合物经减压浓缩,去除溶剂。残余物溶于乙酸乙酯(60ml)中,且经饱和氯化钠水溶液洗涤(50ml×2)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(二氯甲烷:甲醇=0~5%),获得黄色固体的2-氯-4-(1-甲基-1h-苯并[d]咪唑-5-基)嘧啶-5-胺(250mg,24%)。
579、lc-ms m/z:260.1(m+1)+。
580、步骤2:4-(1-甲基-1h-苯并[d]咪唑-5-基)-n2-(4-(4-甲基哌嗪-1-基)苯基)嘧啶-2,5-二胺
581、
582、在混于二恶烷(5ml)内的2-氯-4-(1-甲基-1h-苯并[d]咪唑-5-基)嘧啶-5-胺(200mg,0.77mmol,1.00当量)、4-(4-甲基哌嗪-1-基)苯胺(163mg,0.85mmol,1.10当量)及碳酸铯(753mg,2.31mmol,3.00当量)中,加入xanphos-pd-g3(40mg,0.04mol,0.05当量)。混合物以氮气脱气3次,并在氮气气氛中105℃搅拌16小时。反应混合物的lcms分析表明反应完成。混合物经乙酸乙酯稀释(15ml)、饱和氯化钠水溶液洗涤(5ml)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经制备型hplc纯化(a:0.1%的tfa水溶液;b:乙腈),获得红色固体的实施例41(13.0mg,7%)。
583、制备型hplc条件:
584、仪器:岛津lc8ap制备型hplc系统;色谱柱:boston ods(250mm×21.2mm×10μm,);流动相:溶于水/乙腈的0.1% tfa;梯度:乙腈在1分钟至5分钟时间段为5%,在5分钟至20分钟时间段为5%至25%,在20分钟至25分钟时间段为25%;柱温:25℃;检测波长:214/254nm;流速:20ml/min;
585、lc-ms m/z:415.5(m+1)+;
586、1h nmr(400mhz,d6-dmso):δ9.15(s,1h),8.32(s,1h),8.16(s,1h),8.09(t,j=7.2,1h),7.96(d,j=7.6,1h),7.57(d,j=8.4,2h),6.97(d,j=8.4,2h),4.12(s,3h),3.70~3.65(m,2h),3.58~3.55(m,2h),3.25~3.21(m,2h),2.98~2.96(m,2h),2.95(s,3h)。
587、实施例42:
588、4-(1-甲基-1h-苯并[d]咪唑-5-基)-n-(4-(4-甲基哌嗪-1-基)苯基)-5-(三氟甲基)嘧啶-2-胺
589、2-(1-甲基-1h-苯并[d]咪唑-5-基)-n-(4-(4-甲基哌嗪-1-基)苯基)-5-(三氟甲基)嘧啶-4-胺
590、
591、路线图
592、
593、步骤1:4-氯-n-(4-(4-甲基哌嗪-1-基)苯基)-5-(三氟甲基)嘧啶-2-胺和2-氯-n-(4-(4-甲基哌嗪-1-基)苯基)-5-(三氟甲基)嘧啶-4-胺
594、
595、在溶于dmf(10ml)内的2,4-二氯-5-(三氟甲基)嘧啶化合物(1.0g,4.6mmol,1.0当量)中,加入4-(4-甲基哌嗪-1-基)苯胺化合物(881mg,4.6mol,1.0当量)。混合物在氮气气氛中室温搅拌2天。反应混合物的lcms分析表明反应完成。混合物溶于乙酸乙酯(40ml)中,并经卤水洗涤(40ml×3)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(二氯甲烷:甲醇=0~5%),获得黄色固体的4-氯-n-(4-(4-甲基哌嗪-1-基)苯基)-5-(三氟甲基)嘧啶-2-胺化合物与2-氯-n-(4-(4-甲基哌嗪-1-基)苯基)-5-(三氟甲基)嘧啶-4-胺化合物的混合物(700mg,40.9%)。
596、lc-ms m/z:372.1(m+1)+。
597、步骤2:
598、4-(1-甲基-1h-苯并[d]咪唑-5-基)-n-(4-(4-甲基哌嗪-1-基)苯基)-5-(三氟甲基)嘧啶-2-胺与2-(1-甲基-1h-苯并[d]咪唑-5-基)-n-(4-(4-甲基哌嗪-1-基)苯基)-5-(三氟甲基)嘧啶-4-胺
599、
600、在混于二恶烷/水(10ml/3ml)内的4-氯-n-(4-(4-甲基哌嗪-1-基)苯基)-5-(三氟甲基)嘧啶-2-胺化合物与2-氯-n-(4-(4-甲基哌嗪-1-基)苯基)-5-(三氟甲基)嘧啶-4-胺化合物(700mg,1.88mmol,1.0当量)、(1-甲基-1h-苯并[d]咪唑-5-基)硼酸化合物(530mg,1.88mmol,1.0当量)及碳酸钠(600mg,5.7mmol,3.0当量)中,加入pd(dppf)cl2(100mg,0.1mmol,0.05当量)。混合物以氮气脱气3次,并在氮气气氛中100℃搅拌14小时。反应混合物的lcms分析表明反应完成。混合物以硅藻土过滤,并经减压浓缩,获得粗产物。粗产物经制备型hplc纯化,获得具有峰1(69.9mg,8.0%)和峰2(31.5mg,3.6%)的实施例42,为两种棕色固体。
601、制备型hplc条件:
602、方法a:仪器:岛津lc8ap制备型hplc系统;色谱柱:boston ods(250mm×21.2mm×10μm,);流动相:溶于水/乙腈的0.1% tfa;梯度:乙腈在1分钟至5分钟时间段为5%,在5分钟至20分钟时间段为5%至25%,在20分钟至25分钟时间段为25%;柱温:25℃;检测波长:214/254nm;流速:20ml/min。
603、峰1:
604、lc-ms m/z:468.5(m+1)+;
605、1h nmr(400mhz,cd3od):δ9.15(s,1h),8.73(s,1h),8.01(s,1h),7.94(d,j=8.8hz,1h),7.81(d,j=8.8hz,1h),7.62(d,j=8.4hz,2h),6.98(d,j=7.6hz,2h),4.12(s,3h),3.75~3.72(m,2h),3.59~3.56(m,2h),3.23~3.20(m,2h),2.99~2.95(m,2h),2.94(s,3h)。
606、峰2:
607、lc-ms m/z:468.5(m+1)+;
608、1h nmr(400mhz,cd3od):δ9.06(s,1h),8.69(s,1h),8.64(s,1h),8.51(d,j=8.8hz,1h),7.83(d,j=8.8hz,1h),7.50(d,j=8.4hz,2h),7.12(d,j=8.4hz,2h),4.07(s,3h),3.89~3.86(m,2h),3.65~3.61(m,2h),3.33~3.29(m,2h),3.12~3.08(m,2h),2.98(s,3h)。
609、实施例43:
610、5-氯-4-(异喹啉-7-基)-n-(4-吗啉基苯基)嘧啶-2-胺
611、
612、路线图
613、
614、步骤1:异喹啉-7-基硼酸
615、
616、-78℃下,在混于乙醚(100ml)内的7-溴异喹啉化合物(7g,33.8mmol,1.0当量),逐滴加入正丁基锂(14.8ml,1.1当量)。混合物在氮气气氛中-78℃搅拌0.5小时后,逐滴加入硼酸三异丙酯(b(oi-pr)3)(9.4g,50.7mmol,1.5当量)。混合物在氮气气氛中-78℃搅拌0.5小时后,在25℃下继续搅拌0.5小时。反应混合物的tlc分析表明完全转化至所需产物。反应以盐酸水溶液(2n,6ml)淬灭。在将黄色固体沉淀过滤后,残余物经减压浓缩,获得无色固体的异喹啉-7-基硼酸化合物(5.0g,86.2%)。
617、lc-ms m/z:174.1(m+1)+。
618、步骤2:7-(2,5-二氯嘧啶-4-基)异喹啉
619、
620、在混于thf(50ml)和水(50ml)内的异喹啉-7-基硼酸化合物(3g,16.38mmol,1.0当量)、2,4,5-三氯嘧啶化合物(4.1g,19.65mmol,1.2当量)及碳酸钠(5.11g,19.2mmol,3.0当量)中,加入乙酸钯(90mg,0.33mol,0.02当量)及pph3(180mg,0.66mol,0.04当量)。混合物以氮气脱气3次,并在氮气气氛中80℃搅拌4小时。反应混合物的lcms分析表明反应完成。混合物溶于乙酸乙酯(40ml)中,并经饱和氯化钠水溶液洗涤(40ml×3)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(石油醚:乙酸乙酯:=0~10,加有20%的二氯甲烷),获得黄色固体的7-(2,5-二氯嘧啶-4-基)异喹啉化合物(1g,21.0%)。
621、lc-ms m/z:266.2(m+1)+。
622、步骤3:5-氯-4-(异喹啉-7-基)-n-(4-吗啉基苯基)嘧啶-2-胺
623、
624、在混于异丙醇(3ml)内的7-(2,5-二氯嘧啶-4-基)异喹啉化合物(50mg,0.18mmol,1.0当量)及4-吗啉基苯胺化合物(65mg,0.36mmol,2.0当量)中,加入tfa(2mg,0.018mol,0.1当量)。混合物以氮气脱气3次,并在氮气气氛中于密封管内100℃搅拌16小时。反应混合物的tlc分析表明反应完成。混合物溶于乙酸乙酯(10ml)中,并经饱和碳酸氢钠水溶液洗涤(5ml×3)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经制备型hplc纯化,获得黄色固体的实施例43化合物(8.4mg,13.1%)。
625、制备型hplc条件:
626、方法a:仪器:岛津lc8ap制备型hplc系统;色谱柱:boston ods(250mm×21.2mm×10μm,);流动相:溶有0.1%甲酸的水/乙腈;梯度:乙腈在1分钟至5分钟时间段为5%,在5分钟至20分钟时间段为5%至25%,在20分钟至25分钟时间段为25%;柱温:25℃;检测波长:214/254nm;流速:20ml/min;
627、lc-ms m/z:418.2(m+1)+;
628、1h nmr(400mhz,d6-dmso):δ9.78(s,1h),9.45(s,1h),8.64~8.53(m,3h),8.11(s,2h),7.90(d,j=5.6hz,1h),7.57(d,j=9.2hz,2h),6.88(d,j=9.2hz,2h),3.72~3.69(m,4h),3.01~2.99(m,4h)。
629、实施例44:
630、4-(异喹啉-7-基)-2-((4-吗啉基苯基)氨基)嘧啶-5-腈
631、
632、将悬于二甲基乙酰胺(dma)(10ml)中的5-氯-4-(异喹啉-7-基)-n-(4-吗啉基苯基)嘧啶-2-胺化合物(406mg,2.0mmol,1.0当量)、氰化锌(451mg,3.0mmol,3.0当量)、锌粉(6.5mg,0.1mmol,0.1当量)及xphos-pd-g2(79mg,0.1mmol,0.1当量)以氮气脱气2分钟。混合物在biotage上140℃搅拌1.5小时。反应混合物的lcms分析表明完全转化至所需产物。混合物经过滤及减压浓缩,获得粗产物4-(异喹啉-7-基)-2-((4-吗啉基苯基)氨基)嘧啶-5-腈(500mg,100%)。随后,200mg的粗产物经制备型hplc纯化,获得黄色油状物的实施例44(66.3mg,33.1%)。
633、制备型hplc条件:
634、方法a:仪器:岛津lc8ap制备型hplc系统;色谱柱:boston ods(250mm×21.2mm×10μm,);流动相:溶有0.1%甲酸的水/乙腈;梯度:乙腈在1分钟至5分钟时间段为5%,在5分钟至20分钟时间段为5%至25%,在20分钟至25分钟时间段为25%;柱温:25℃;检测波长:214/254nm;流速:20ml/min;
635、lc-ms m/z:409.3(m+1)+;
636、1h nmr(400mhz,d6-dmso)δ9.47(s,1h),8.95(s,1h),8.69(s,1h),8.62(d,j=5.6hz,1h),8.46(s,1h),8.22(s,1h),8.18(s,1h),7.94(s,1h),7.60(m,2h),6.94(d,j=8.8hz,2h),3.71(m,4h),3.04(m,4h)。
637、实施例45:
638、4-(异喹啉-7-基)-2-((4-吗啉基苯基)氨基)嘧啶-5-羧酸
639、
640、在混于乙酸(1ml)内的4-(异喹啉-7-基)-2-((4-吗啉基苯基)氨基)嘧啶-5-腈化合物(200mg,0.49mmol,1.0当量)中,加入硫酸/水(2.5ml,4:1)。混合物以氮气脱气3次,并在氮气气氛中90℃搅拌16小时。反应混合物的lcms分析表明完全转化至所需产物。混合物以碳酸氢钠淬灭,随后以dcm(3×15ml)萃取。合并有机层经无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经制备型hplc纯化,获得黄色固体的实施例45化合物(13.1mg,6.6%)。
641、制备型hplc条件:
642、方法a:仪器:岛津lc8ap制备型hplc系统;色谱柱:boston ods(250mm×21.2mm×10μm,);流动相:溶有0.1%甲酸的水/乙腈;梯度:乙腈在1分钟至5分钟时间段为5%,在5分钟至20分钟时间段为5%至25%,在20分钟至25分钟时间段为25%;柱温:25℃;检测波长:214/254nm;流速:20ml/min;
643、lc-ms m/z:427.3(m+1)+;
644、1h nmr(400mhz,d6-dmso):δ9.84(s,1h),9.38(s,1h),8.58(s,1h),8.55(d,j=6.0hz,1h),8.39(s,1h),8.03(d,j=8.8hz,1h),7.97(d,j=8.8hz,1h),7.88(s,2h),7.63(d,j=8.0hz,2h),7.46(s,1h),6.90(d,j=8.4hz,2h),3.75~3.68(s,4h),3.03~2.99(s,4h)。
645、实施例46:
646、4-(异喹啉-7-基)-2-((4-吗啉基苯基)氨基)嘧啶-5-羧酰胺
647、
648、在溶于dmso(3ml)内的4-(异喹啉-7-基)-2-((4-吗啉基苯基)氨基)嘧啶-5-腈化合物(200mg,0.5mmol,1.0当量)中,缓慢加入2n氢氧化钠(5ml)及过氧化氢(1.25ml,10mmol,20当量)。混合物在25℃下搅拌16小时。反应混合物的lcms分析表明反应完成。混合物以水稀释(15ml),并以dcm萃取(3×15ml)。合并有机层经无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经制备型hplc纯化,获得黄色固体的实施例46化合物(21.4mg,10.7%)。
649、制备型hplc条件:
650、方法b:仪器:岛津lc8ap制备型hplc系统;色谱柱:boston ods(250mm×21.2mm×10μm,);流动相:溶有0.1%甲酸的水/乙腈;梯度:乙腈在1分钟至5分钟时间段为5%,在5分钟至20分钟时间段为10%至30%,在20分钟至25分钟时间段为30%;柱温:25℃;检测波长:214/254nm;流速:20ml/min;
651、lc-ms m/z:427.4(m+1)+;
652、1h nmr(400mhz,d6-dmso):δ9.84(s,1h),9.39(s,1h),8.58(s,1h),8.55(d,j=5.6hz,1h),8.39(s,1h),8.03(d,j=8.4hz,1h),7.97(d,j=8.4hz,1h),7.87(d,j=5.6hz,2h),7.64(d,j=8.8hz,2h),7.46(s,1h),6.90(d,j=8.8hz,2h),3.72~3.69(m,4h),3.05~3.01(m,4h)。
653、实施例47:
654、5-氯-n-(4-(4-甲基哌嗪-1-基)苯基)-4-(喹唑啉-7-基)嘧啶-2-胺
655、
656、路线图
657、
658、步骤1:7-(2,5-二氯嘧啶-4-基)喹唑啉
659、
660、在混于无水二恶烷(15ml)内的7-溴喹唑啉化合物(200.0mg,0.95mmol,1.0当量)、b2pin2(291.5mg,1.15mmol,1.2当量)及乙酸钾(281.7mg,1.91mmol,3.0当量)中,加入pd(dppf)cl2(40.0mg,0.05mmol,0.05当量)。混合物以氮气脱气3次,并在氮气气氛中110℃搅拌3小时。反应混合物的lcms分析表明反应完成,并加入2,4,5-三氯嘧啶化合物(350.9mg,1.91mmol,2.0当量)。混合物以氮气脱气3次,并在氮气气氛中100℃搅拌3小时。反应混合物的lcms分析表明反应完成。混合物经过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(二氯甲烷:甲醇=0~1),获得粉红色固体的7-(2,5-二氯嘧啶-4-基)喹唑啉化合物(250.0mg,94.0%)。
661、lc-ms m/z:277.1(m+1)+。
662、步骤2:5-氯-n-(4-(4-甲基哌嗪-1-基)苯基)-4-(喹唑啉-7-基)嘧啶-2-胺
663、
664、在混于无水二恶烷(15ml)内的7-(2,5-二氯嘧啶-4-基)喹唑啉化合物(200mg,0.722mmol,1.0当量)、4-(4-甲基哌嗪-1-基)苯胺化合物(138.1mg,0.722mmol,1.0当量)及碳酸铯(325.8mg,2.166mmol,3.0当量)中,加入xantphos-pd-g3(60.0mg,0.062mmol,0.1当量)。混合物以氮气脱气3次,并在氮气气氛中110℃搅拌10小时。反应混合物的lcms分析表明反应完成。混合物经过滤及减压浓缩,获得粗产物。粗产物经制备型hplc纯化,获得黄色固体的实施例47化合物(31.0mg,7.8%)。
665、制备型hplc条件:
666、方法a:仪器:岛津lc8ap制备型hplc系统;色谱柱:boston ods(250mm×21.2mm×10μm,);流动相:溶于水/乙腈的0.1% tfa;梯度:乙腈在1分钟至5分钟时间段为5%,在5分钟至20分钟时间段为5%至25%,在20分钟至25分钟时间段为25%;柱温:25℃;检测波长:214/254nm;流速:20ml/min;
667、lc-ms m/z:432.1(m+1)+;
668、1h nmr(400mhz,cd3od):δ8.65(s,1h),8.46(s,1h),7.93(d,j=8.0hz,1h),7.79(s,1h),7.68(s,1h),7.59(d,j=8.8hz,2h),6.97(d,j=8.8hz,2h),6.25~6.21(m,2h),3.74~3.70(m,2h),3.59~3.56(m,2h),3.25~3.19(m,2h),2.99~2.95(m,2h),2.94(s,3h)。
669、实施例48:
670、5-氯-n-(2-氟-6-甲氧基苯基)-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺
671、
672、路线图
673、
674、步骤1:5-氯-n-(2-氟-6-甲氧基苯基)-2-(甲硫基)嘧啶-4-羧酰胺
675、
676、在溶于dmf(25ml)内的5-氯-2-(甲硫基)嘧啶-4-羧酸化合物(1.5g,7.33mmol,1.0当量)中,加入2-氟-6-甲氧基苯胺化合物(1.03g,7.33mmol,1.0当量)、2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(hatu)(4.18g,11.0mmol,1.5当量)及n,n-二异丙基乙基胺(diea)(2.8g,22.0mmol,3.0当量)。混合物在氮气气氛中室温搅拌16小时。反应混合物的lcms分析表明完全转化至所需产物。混合物减压浓缩后,残余物以水(50ml)和dcm(50ml×2)萃取。有机层经无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(甲醇:二氯甲烷=0~1%),获得黄色固体的5-氯-n-(2-氟-6-甲氧基苯基)-2-(甲硫基)嘧啶-4-羧酰胺化合物(1.4g,58%,90% tlc纯度)。
677、lc-ms m/z:328.1(m+1)+。
678、步骤2:5-氯-n-(2-氟-6-甲氧基苯基)-2-(甲磺酰基)嘧啶-4-羧酰胺
679、
680、0℃下,在悬于dcm(20ml)内的5-氯-n-(2-氟-6-甲氧基苯基)-2-(甲硫基)嘧啶-4-羧酰胺化合物(1g,3.0mmol,1.0当量)中,加入间氯过氧苯甲酸(mcpba)(2.11g,12.0mmol,4.0当量)。混合物在氮气气氛中室温搅拌4小时。反应混合物的lcms分析表明反应完成。混合物以碳酸氢钠水溶液碱化至ph=8,并以dcm萃取(50ml×2)。合并有机层经饱和氯化钠水溶液洗涤(50ml×2)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(甲醇:二氯甲烷=0~5%),获得白色固体的5-氯-n-(2-氟-6-甲氧基苯基)-2-(甲磺酰基)嘧啶-4-羧酰胺化合物(710mg,64%)。
681、lc-ms m/z:360.1(m+1)+。
682、步骤2:5-氯-n-(2-氟-6-甲氧基苯基)-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺
683、
684、在溶于dma(8ml)的5-氯-n-(2-氟-6-甲氧基苯基)-2-(甲磺酰基)嘧啶-4-羧酰胺化合物(250mg,0.7mmol,1.0当量)、4-(4-甲基哌嗪-1-基)苯胺(266mg,1.4mmol,2.0当量)及diea(361mg,2.8mmol,4.0当量)中。混合物在微波反应器内130℃搅拌1小时。混合物经减压浓缩,获得粗产物。粗产物溶于dmf/甲酸(3ml,10:1)中,并经制备型hplc纯化(a:0.1%甲酸水溶液;b:乙腈),获得黄色固体的
685、实施例48化合物(17.4mg,5.3%)。
686、制备型hplc条件:
687、方法a:仪器:岛津lc8ap制备型hplc系统;色谱柱:boston ods(250mm×21.2mm×10μm,);流动相:溶有0.1%盐酸的水/乙腈;梯度:乙腈在1分钟至5分钟时间段为5%,在5分钟至20分钟时间段为5%至25%,在20分钟至25分钟时间段为25%;柱温:25℃;检测波长:214/254nm;流速:20ml/min;
688、lc-ms m/z:471.4(m+1)+;
689、1h nmr(400mhz,dmso):δ8.52(s,1h),7.63(d,j=8.4hz,2h),7.31(q,j=8.0hz,1h),7.03(d,j=8.8hz,2h),6.92(d,j=8.4hz,1h),6.83(t,j=8.8hz,1h),3.88(s,3h),3.78(d,j=12.4hz,2h),3.60(d,j=12.0hz,2h),3.30(s,2h),3.04(t,j=12.0hz,2h),2.96(s,3h)。
690、实施例49:
691、5-氯-n-(2-氯-6-甲氧基苯基)-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺
692、
693、路线图
694、
695、步骤1:5-氯-2-(甲硫基)嘧啶-4-羧酸甲酯
696、
697、0℃下,在混于甲醇(300ml)内的5-氯-2-(甲硫基)嘧啶-4-羧酸(30g,15mmol,1.0当量)中,缓慢逐滴加入氯化亚砜(100ml)。混合物以氮气脱气3次,并在氮气气氛中室温搅拌16小时。反应混合物的lcms分析表明完全转化至所需产物。混合物经减压浓缩,去除多余的溶剂。残余物经硅胶柱层析纯化(石油醚:乙酸乙酯=0~20%),获得淡黄色固体的5-氯-2-(甲硫基)嘧啶-4-羧酸甲酯(28g,86.2%)。
698、lc-ms m/z:219.4(m+1)+。
699、步骤2:5-氯-2-(甲磺酰基)嘧啶-4-羧酸甲酯
700、
701、0℃下,在混于二氯甲烷(300ml)内的5-氯-2-(甲硫基)嘧啶-4-羧酸甲酯(33g,15.1mmol,1.0当量)中,分数批加入间氯过氧苯甲酸(104g,60.3mmol)。混合物在氮气气氛中室温搅拌4小时。反应混合物的lcms分析表明完全转化至所需产物。混合物经减压浓缩,去除多余的溶剂。残余物经硅胶柱层析纯化(石油醚:乙酸乙酯=0~33%),获得淡黄色固体的5-氯-2-(甲磺酰基)嘧啶-4-羧酸甲酯(31g,82.0%)。
702、lc-ms m/z:251.5(m+1)+。
703、步骤3:n-(4-(4-甲基哌嗪-1-基)苯基)甲酰胺
704、
705、将混于甲酸(100ml)内的4-(4-甲基哌嗪-1-基)苯胺(8.0g,41.7mmol,1.0当量)在氮气气氛中100℃下搅拌16小时。反应混合物的lcms分析表明完全转化至所需产物。混合物经减压浓缩,去除多余的溶剂,获得淡黄色固体的粗产化合物n-(4-(4-甲基哌嗪-1-基)苯基)甲酰胺(13.1g,100%),该粗产物直接用于下一步骤,无需进一步纯化。
706、lc-ms m/z:220.3(m+1)+;
707、步骤4:5-氯-2-(n-(4-(4-甲基哌嗪-1-基)苯基)甲酰胺基)嘧啶-4-羧酸甲酯
708、
709、0℃下,在混于dmf(50ml)内的n-(4-(4-甲基哌嗪-1-基)苯基)甲酰胺化合物(2.2g,10.0mmol,1.0当量)中,分数批加入氢化钠(1.2g,30.0mmol,3.0当量)。搅拌0.5小时后,加入5-氯-2-(甲磺酰基)嘧啶-4-羧酸甲酯(2.5g,10.0mmol,1.0当量)。混合物在氮气气氛中室温搅拌4小时。反应混合物的lcms分析表明完全转化至所需产物。混合物以饱和氯化铵(0.5ml)淬灭,并经减压浓缩,去除多余的溶剂。残余物经硅胶柱层析纯化(二氯甲烷:甲醇=0~5%),获得淡黄色固体的5-氯-2-(n-(4-(4-甲基哌嗪-1-基)苯基)甲酰胺基)嘧啶-4-羧酸甲酯化合物(3.1g,53.8%)。
710、lc-ms m/z:390.6(m+1)+。
711、步骤5:5-氯-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-羧酸
712、
713、在混于thf(20ml)内的5-氯-2-(n-(4-(4-甲基哌嗪-1-基)苯基)甲酰胺基)嘧啶-4-羧酸甲酯(2.1g,10.0mmol,1.0当量)中,加入4n的盐酸(20ml)。混合物在氮气气氛中90℃搅拌16小时。反应混合物的lcms分析表明完全转化至所需产物。混合物冷却至室温后,对悬浮液进行过滤。滤饼经真空干燥,获得淡黄色固体的5-氯-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-羧酸(1.1g,58.8%),该化合物直接用于下一步骤,无需进一步纯化。
714、lc-ms m/z:348.3(m+1)+。
715、步骤6:5-氯-n-(2-氯-6-甲氧基苯基)-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺
716、
717、在混于吡啶内的5-氯-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-羧酸(80mg,0.231mmol,1.0当量)及2-氯-6-甲氧基苯胺(40mg,0.254mmol,1.1当量)中,加入三氯氧磷(5滴)。混合物以氮气脱气3次,并在氮气气氛中室温搅拌2小时。反应混合物的lcms分析表明完全转化至所需产物。反应混合物经过滤和减压浓缩,获得粗产物。粗产物经制备型hplc纯化,获得淡黄色固体的实施例49(5.7mg,5%)。制备型hplc条件:
718、方法a:仪器:岛津lc8ap制备型hplc系统;色谱柱:boston ods(250mm×21.2mm×10μm,);流动相:溶有0.1%盐酸的水/乙腈;梯度:乙腈在1分钟至5分钟时间段为5%,在5分钟至20分钟时间段为5%至25%,在20分钟至25分钟时间段为25%;柱温:25℃;检测波长:214/254nm;流速:20ml/min;
719、lc-ms m/z:487.4(m+1)+;
720、1h nmr(400mhz,cd3od):δ8.49(s,1h),7.58(d,j=8.4hz,2h),7.28(d,j=8.0hz,1h),7.09(d,j=8.0hz,1h),7.04(d,j=8.4hz,1h),6.95(d,j=8.4hz,2h),3.86(s,3h),3.24~3.19(m,4h),2.83~2.79(m,4h),2.48(s,3h)。
721、
722、
723、实施例54:
724、5-氯-n-(2-氟-6-甲氧基苯基)-n-甲基-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)异烟酰胺
725、
726、路线图
727、
728、步骤1:2,5-二氯-n-(2-氟-6-甲氧基苯基)异烟酰胺
729、
730、0℃下,在氮气气氛中,在混于dmf(30ml)内的2,5-二氯异烟酸化合物(2.0g,10.4mmol,1.0当量)中,分批加入2-氟-6-甲氧基苯胺化合物(1.5g,10.4mmol,1.0当量)、hatu(4.7g,12.5mmol,1.2当量)及diea(2.7g,20.8mmol,2.0当量)。室温搅拌4小时后,反应混合物的lcms分析表明完全转化至所需产物。反应混合物以水稀释(30ml),并以乙酸乙酯萃取(20ml×2)。有机层经饱和氯化钠水溶液洗涤(20ml×2)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(石油醚:乙酸乙酯=0~3%),获得黄色固体的2,5-二氯-n-(2-氟-6-甲氧基苯基)异烟酰胺化合物(1.5g,46%)。
731、lc-ms m/z:315.3(m+1)+。
732、步骤2:2,5-二氯-n-(2-氟-6-甲氧基苯基)-n-甲基异烟酰胺
733、
734、0℃下,在混于dmf(20ml)内的2,5-二氯-n-(2-氟-6-甲氧基苯基)异烟酰胺化合物(1.1g,3.5mmol,1.0当量)中,加入氢化钠(280mg,7.0mmol,2.0当量)。混合物在氮气气氛中0℃搅拌0.5小时。随后,加入碘甲烷(995mg,7.0mmol,2.0当量)。混合物在氮气气氛中室温搅拌1小时。反应混合物的lcms分析表明完全转化至所需产物。随后,混合物以水(20ml)淬灭,并以乙酸乙酯萃取(20ml×2)。有机层经饱和氯化钠水溶液洗涤(20ml×2)、无水硫酸钠干燥、过滤及减压浓缩,获得粗产物。粗产物经硅胶柱层析纯化(石油醚:乙酸乙酯=0~3%),获得白色固体的2,5-二氯-n-(2-氟-6-甲氧基苯基)-n-甲基异烟酰胺化合物(1.0g,87%)。
735、lc-ms m/z:329.3(m+1)+。
736、步骤3:5-氯-n-(2-氟-6-甲氧基苯基)-n-甲基-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)异烟酰胺
737、
738、在氮气气氛中,在混于二恶烷(5ml)内的2,5-二氯-n-(2-氟-6-甲氧基苯基)-n-甲基异烟酰胺化合物(250mg,0.76mmol,1.0当量)及化合物4-(4-甲基哌嗪-1-基)苯胺(146mg,0.76mmol,1.0当量)中,加入碳酸铯(497mg,1.52mmol,2.0当量)、2,2'-双(二苯膦基)-1,1'-联萘(binap)(40mg,0.05mmol,0.1当量)及pd2(dba)3(35mg,0.05mmol,0.1当量)。在氮气气氛中,混合物在微波反应器中140℃搅拌1.5小时。反应混合物的lcms分析表明完全转化至所需产物。随后,混合物以水稀释(20ml),并以乙酸乙酯萃取(20ml×2)。有机层经无水硫酸钠干燥、过滤及减压浓缩。残余物经制备型hplc纯化,获得红色固体的实施例54(140mg,38%)。
739、制备型hplc条件:
740、方法a:仪器:岛津lc8ap制备型hplc系统;色谱柱:boston ods(250mm×21.2mm×10μm,);流动相:溶有0.1%甲酸的水/乙腈;梯度:乙腈在1分钟至5分钟时间段为5%,在5分钟至20分钟时间段为5%至25%,在20分钟至25分钟时间段为25%;柱温:25℃;检测波长:214/254nm;流速:20ml/min;
741、lc-ms m/z:484.1(m+1)+;
742、1h nmr(400mhz,d6-dmso):δ8.92(s,1h),7.93(s,1h),7.22(d,j=8.8,2h),6.89(d,j=8.8,2h),6.81(d,j=8.8,2h),6.44(s,1h),3.71(s,3h),3.33(s,3h),2.90(s,4h),2.50(d,j=11.6,4h)。
743、实施例55:
744、n-(5-氯-2-((2-异丙基-7-甲氧基-1,2,3,4-四氢异喹啉-6-基)氨基)嘧啶-4-基)-2-氟-6-甲氧基苯甲酰胺
745、
746、路线图
747、
748、步骤1:(4-甲氧基苯乙基)氨基甲酸异丁酯
749、
750、0℃下,在溶于dcm(50ml)内的2-(4-甲氧基苯基)乙-1-胺(5g,33.07mmol,1当量)中,加入碳酸钠(7.01g,66.13mmol,2当量)。随后,在0℃下,将氯甲酸异丁酯(5.42g,39.68mmol,1.2当量)缓慢加入反应物中。完成后,所得混合物在0℃下搅拌2小时,获得白色悬浮液。lcms表明反应完成。反应物以水稀释,并以dcm萃取(30ml×3)。合并有机层经卤水洗涤、硫酸钠干燥、过滤及浓缩,获得白色固体的标题所述产物(8.2g,收率98.68%)。该产物直接用于下一步骤,无需进一步纯化。
751、lc-ms m/z:252.2(m+1)+。
752、步骤2:7-甲氧基-3,4-二氢异喹啉-1(2h)-酮
753、
754、140℃下,将(4-甲氧基苯乙基)氨基甲酸异丁酯(8.2g,32.63mmol,1当量)分批加入处于搅拌状态的多聚磷酸溶液(40ml)中。所得混合物在140℃下搅拌1小时,以获得棕色溶液。lcms表明反应完成。反应以水稀释,并以dcm萃取(30ml×3)。合并有机层经卤水洗涤、硫酸钠干燥、过滤及浓缩,获得黄色油状物的标题所示产物(6.1g,收率105.54%,粗产物),该产物直接用于下一步骤,无需进一步纯化。
755、lc-ms m/z:178.1(m+1)+。
756、步骤3:7-甲氧基-6-硝基-3,4-二氢异喹啉-1(2h)-酮
757、
758、0℃下,在溶于乙腈(20ml)和三氟乙酸酐(20ml)内的7-甲氧基-3,4-二氢异喹啉-1(2h)-酮(5.6g,31.60mmol,1当量)中,分批加入14.8m的硝酸(1.92ml,28.44mmol,0.9当量)。所得混合物在0℃下搅拌1小时,获得黑棕色溶液。lcms表明起始物质已完全消耗且检测到一个具有所需物质的主峰。反应物以碳酸氢钠水溶液中和,并以dcm萃取(40ml×3)。合并有机层经卤水洗涤、硫酸镁干燥、过滤及浓缩,获得棕色油状物的标题所示产物(5.89g,收率84.26%),该产物直接用于下一步骤,无需进一步纯化。
759、lc-ms m/z:223.1(m+1)+。
760、步骤4:7-甲氧基-6-硝基-1,2,3,4-四氢异喹啉
761、
762、0℃下,在溶于thf(20ml)内的7-甲氧基-6-硝基-3,4-二氢异喹啉-1(2h)-酮(6.3g,28.48mmol,1当量)中,逐滴加入1m的硼烷(85.45ml,85.45mmol,3当量)。完成后,所得混合物在90℃下搅拌2小时,以获得棕色溶液。lcms表明起始物质已大部分消耗。反应物在90℃下继续搅拌4小时。lcms表明起始物质已完全消耗。反应以0℃的甲醇淬灭,混合物经浓缩,去除大部分的溶剂。所得混合物加入1n的盐酸(30ml),且在90℃下搅拌2小时,以获得棕色溶液。lcms表明检测到一个具有所需物质的主峰。反应物以水稀释,并以dcm洗涤(30ml×2)。水层以浓碳酸钠碱化至ph=8~9,并以dcm萃取(30ml×3)。合并有机层经卤水洗涤、硫酸镁干燥、过滤及浓缩,获得棕色油状物的标题所示产物(1.9g,收率32.20%)。该产物用于下一步骤,无需进一步纯化。
763、lc-ms m/z:209.1(m+1)+。
764、步骤5:2-异丙基-7-甲氧基-6-硝基-1,2,3,4-四氢异喹啉
765、
766、在溶于乙腈(30ml)内的7-甲氧基-6-硝基-1,2,3,4-四氢异喹啉(1.9g,9.13mmol,1当量)中,加入碳酸钾(5.04g,36.50mmol,4当量)及2-碘丙烷(7.76g,45.63mmol,5当量)。完成后,所得混合物在65℃下搅拌3小时,获得棕色悬浮液。lcms表明起始物质已大部分消耗且检测到一个具有所需物质的峰。反应物经浓缩,去除大部分的溶剂。残余物以水稀释,并以乙酸乙酯洗涤(10ml×3)。水层以1n的盐酸酸化至ph=5~6,并以乙酸乙酯萃取(20ml×3)。合并有机层经卤水洗涤、硫酸钠干燥及过滤,并浓缩至干态。残余物经快速柱纯化(二氧化硅,以100%的dcm至溶有5%甲醇的dcm以及0.5%的氨水洗脱),获得棕色油状物的标题所示产物(1.63g,收率71.49%)。
767、lc-ms m/z:251.5(m+1)+。
768、步骤6:2-异丙基-7-甲氧基-1,2,3,4-四氢异喹啉-6-胺
769、
770、氮气气氛中,在溶于甲醇(20ml)内的2-异丙基-7-甲氧基-6-硝基-1,2,3,4-四氢异喹啉(1.63g,6.51mmol,1当量)中,加入钯碳(0.3g)。所得混合物以氢气脱气数次,并在60psi下20℃搅拌16小时,获得黑色悬浮液。tlc表明起始物质已完全消耗且检测到一个主斑。反应物以硅藻土垫过滤,且滤过物浓缩至干态。残余物经冻干,获得棕色油状物的标题所示产物(1.37g,收率95.80%)。该不纯的产物(约1g)经快速柱纯化(二氧化硅,以100%的乙酸乙酯至溶有3%甲醇的乙酸乙酯洗脱),获得棕色油状物的标题所示产物(497mg)。
771、lc-ms m/z:221.3(m+1)+。
772、步骤7:2-氟-6-甲氧基苯甲酰氯
773、
774、0℃下,在溶于dcm(10ml)内的2-氟-6-甲氧基苯甲酸(1g,5.88mmol,1当量)中,加入乙二酰氯(37.73g,29.39mmol,5当量)及dmf(4mg,0.06mmol,0.01当量)。反应物在26℃下搅拌1小时,获得淡黄色溶液。tlc(以甲醇淬灭)表明起始物质已完全消耗且检测到一个新的斑点。反应浓缩至干态后,残余物以dcm(20ml×3)进行交换,获得浅黄色胶状物的标题所示产物(1.1g)。该产物用于下一步骤,无需进一步纯化。
775、步骤8:n-(2,5-二氯嘧啶-4-基)-2-氟-6-甲氧基苯甲酰胺
776、
777、0℃下,在溶于dmf(7ml)内的2,5-二氯嘧啶-4-胺(0.96g,5.83mol,1当量)中,加入氢化钠(0.35g,5.83mmol,1.5当量)。搅拌0.5小时后,在0℃下,向反应物中逐滴加入溶于dmf(3ml)内的2-氟-6-甲氧基苯甲酰氯(1.1g,5.83mmol,1当量)。反应物在26℃下搅拌16小时,获得黄色悬浮液。lcms表明反应进行状况良好。反应物以水稀释后,混合物以1n盐酸水溶液酸化至ph=5~6,且以乙酸乙酯萃取。合并有机层经氯化铵水溶液和卤水洗涤、硫酸钠干燥及过滤后,浓缩至干态。残余物经快速柱纯化(二氧化硅,以溶有5%~20%乙酸乙酯的石油醚洗脱),获得米白色固体的标题所示产物(1.21g,收率65.76%)。
778、lc-ms m/z:315.9(m+1)+。
779、步骤9:n-(5-氯-2-((2-异丙基-7-甲氧基-1,2,3,4-四氢异喹啉-6-基)氨基)嘧啶-4-基)-2-氟-6-甲氧基苯甲酰胺
780、
781、氮气气氛中,在溶于二恶烷(1ml)内的n-(2,5-二氯嘧啶-4-基)-2-氟-6-甲氧基苯甲酰胺(50mg,158.17μmol,1当量)及2-异丙基-7-甲氧基-1,2,3,4-四氢异喹啉-6-胺(52.27mg,237.26μmol,1.5当量)中,加入xantphos(18.30mg,31.63μmol,0.2当量)、碳酸铯(154.61mg,474.51μmol,3当量)及乙酸钯(3.55mg,15.82μmol,0.1当量)。所得混合物在100℃下微波搅拌0.5小时,获得棕色悬浮液。lcms表明起始物质已大部分消耗且检测到一个具有所需物质的主峰。混合物在100℃下继续微波搅拌0.5小时。lcms表明反应混乱,且检测到一个具有所需物质的峰。反应物以dcm/甲醇=10/1稀释,并以硅藻土垫过滤。滤过物浓缩后,残余物经快速柱纯化(二氧化硅,以溶有30%~100%乙酸乙酯的石油醚洗脱)。流出物收集后,经浓缩和冻干,获得淡黄色固体的实施例55(19.5mg,收率6.16%)。
782、lc-ms m/z:500.4(m+1)+;
783、1h nmr(400mhz,dmso-d6):δ10.75(s,1h),8.44(s,1h),8.10(s,1h),7.76(s,1h),7.44(q,j=8.0hz,1h),6.94(d,j=8.5hz,1h),6.87(t,j=8.7hz,1h),6.72(s,1h),3.79(s,3h),3.78(s,3h),3.61(s,2h),2.85(s,2h),2.70(br,5h),1.07(s,3h),1.05(s,3h)。
784、
785、
786、
787、实施例63:
788、n-(5-氯-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)-2-氟-6-甲氧基-n-甲基苯甲酰胺
789、
790、路线图
791、
792、步骤1:n-(2,5-二氯嘧啶-4-基)-2-氟-6-甲氧基-n-甲基苯甲酰胺
793、
794、0℃下,在溶于dmf(2ml)内的n-(2,5-二氯嘧啶-4-基)-2-氟-6-甲氧基苯甲酰胺(200mg,632.68μmol,1当量)中,加入氢化钠(37.96mg,949.03mmol,1.5当量)。搅拌0.5小时后,在0℃下,向反应物内加入碘甲烷(449.01mg,3.16mmol,1当量)。反应物在26℃下搅拌16小时,获得棕色悬浮液。lcms表明检测到一个具有所需物质的主峰。反应物以水稀释后,以1n盐酸酸化至ph=2~3,并以dcm洗涤(5ml×2)。水层以碳酸钠水溶液碱化至ph=10~11,并以dcm萃取(5ml×3)。合并有机层经卤水洗涤、硫酸钠干燥及过滤后,浓缩至干态。残余物经快速柱纯化(二氧化硅,以溶有10%~50%乙酸乙酯的石油醚洗脱)。流出物收集后,经浓缩和冻干,获得淡黄色胶状物的标题所示产物(133.5mg,收率63.92%)。
795、lc-ms m/z:330.0(m+1)+。
796、步骤2:n-(5-氯-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)-2-氟-6-甲氧基-n-甲基苯甲酰胺
797、
798、氮气气氛中,在溶于二恶烷(1ml)内的n-(2,5-二氯嘧啶-4-基)-2-氟-6-甲氧基-n-甲基苯甲酰胺(50mg,151.45μmol,1当量)及4-(4-甲基哌嗪-1-基)苯胺(30.42mg,159.02μmol,1.05当量)中,加入xantphos(17.53mg,30.29μmol,0.2当量)、碳酸铯(98.69mg,302.90μmol,2当量)及乙酸钯(3.40mg,15.15μmol,0.1当量)。所得混合物在100℃下微波搅拌0.5小时,获得棕色悬浮液。lcms表明起始物质已完全消耗和且检测到一个具有所需物质的主峰。反应物以dcm/甲醇=10/1稀释,并以硅藻土垫过滤。滤过物浓缩后,残余物经快速柱纯化(二氧化硅,以溶有30%~100%乙酸乙酯的石油醚洗脱)。流出物收集后,经浓缩和冻干,获得黄色固体的实施例63(38.7mg,收率52.69%)。
799、lc-ms m/z:485.6(m+1)+;
800、1h nmr(400mhz,dmso-d6):δ9.53(s,1h),8.44(s,1h),7.47(s,1h),7.26(s,2h),6.85(s,2h),6.73(d,j=34.1hz,2h),3.70(d,j=110.3hz,3h),3.29(s,3h),3.06(t,j=5.1hz,4h),2.47(t,j=5.1hz,4h),2.22(s,3h)。
801、实施例64:
802、2-氯-n-(5-氯-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)-6-甲氧基苯甲酰胺
803、
804、路线图
805、
806、步骤1:2-氯-6-甲氧基苯甲酰氯
807、
808、0℃下,在溶于dcm(3ml)内的2-氯-6-甲氧基苯甲酸(150mg,803.90μmol,1当量)中,加入乙二酰氯(510.15mg,4.02mmol,5当量)及dmf(0.59mg,8.04μmol,0.01当量)。反应物在24℃下搅拌1小时,获得淡黄色溶液。tlc(以甲醇淬灭)表明起始物质已完全消耗且检测到一个新的斑点。反应物浓缩至干态后,残余物以dcm(20ml×3)进行交换,获得浅黄色胶状物的标题所示产物(168mg,收率101.92%)。
809、步骤2:2-氯-n-(2,5-二氯嘧啶-4-基)-6-甲氧基苯甲酰胺
810、
811、0℃下,在溶于dmf(2ml)内的2,5-二氯嘧啶-4-胺(120mg,731.76μmol,1当量)中,加入氢化钠(26.34mg,1.10mmol,1.5当量)。搅拌0.5小时后,在0℃下,向反应物内逐滴加入溶于dcm(2ml)中的2-氯-6-甲氧基苯甲酰氯(165.04mg,804.93mmol,1当量)。反应物在24℃下搅拌2小时,获得黄色悬浮液。lcms表明反应进行状况良好。反应物以水稀释后,混合物以1n盐酸水溶液酸化至ph=5~6,并以dcm萃取。合并有机层经氯化铵水溶液和卤水洗涤、硫酸钠干燥及过滤后,浓缩至干态。残余物经冻干,获得米白色固体的标题所示产物(273mg,收率112.18%)。
812、lc-ms m/z:331.9(m+1)+。
813、步骤3:2-氯-n-(5-氯-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)-6-甲氧基苯甲酰胺
814、
815、氮气气氛中,在溶于二恶烷(1ml)内的2-氯-n-(2,5-二氯嘧啶-4-基)-6-甲氧基苯甲酰胺(50mg,150.35μmol,1当量)及4-(4-甲基哌嗪-1-基)苯胺(28.76mg,150.35μmol,1当量)中,加入xantphos(13.05mg,22.55mol,0.15当量)、碳酸铯(97.97mg,300.69μmol,2当量)及乙酸钯(2.36mg,10.52μmol,0.07当量)。所得混合物在100℃下微波搅拌0.5小时,获得棕色溶液。lcms表明起始物质已完全消耗且检测到一个具有所需物质的主峰。反应物以硅藻土垫过滤,并浓缩至干态。残余物经快速柱纯化(c18,以含有0.01%的tfa且溶有10%~80%乙腈的水洗脱,流速为60ml/min),且流出物经冻干,获得淡黄色固体的实施例64(7.2mg,收率9.83%)。
816、lc-ms m/z:487.6(m+1)+;
817、1h nmr(400mhz,dmso-d6):δ10.76(s,1h),9.66~9.43
818、(m,1h),8.42(s,1h),7.70~7.47(m,2h),7.40(s,1h),7.09(d,j=8.0hz,2h),6.84(d,j=8.6hz,2h),3.78(s,3h),3.05(t,j=4.9hz,4h),2.44(t,j=5.0hz,4h),2.21(s,3h)。
819、
820、实施例66:
821、n-(5-氯-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)-4-甲基烟酰胺
822、
823、路线图2
824、
825、步骤1:4-甲基烟酰氯
826、
827、0℃下,在溶于dcm(30ml)内的4-甲基烟酸化合物(1g,7.29mmol,1当量)中,加入乙二酰氯(4.63g,36.46mmol,5当量)及dmf(53.30mg,0.73mmol,0.1当量)。反应物室温搅拌1小时,获得黄色溶液。tlc(以甲醇淬灭)表明起始物质已完全消耗且检测到一个新的斑点。反应物浓缩至干态后,残余物以dcm(20ml×3)进行交换,获得黄色固体的4-甲基烟酰氯(1.14g,收率:100.9%)。
828、步骤2:n-(2,5-二氯嘧啶-4-基)-4-甲基烟酰胺
829、
830、0℃下,在混于thf(10ml)内的2,5-二氯嘧啶-4-胺(1g,6.10mmol,1当量)中,加入氢化钠(0.37g,9.15mmol,1.5当量)。搅拌0.5小时后,在0℃下向反应物中逐滴加入溶于thf(5ml)和dcm(5ml)中的4-甲基烟酰氯。反应物在室温下搅拌16小时,获得棕色溶液。反应混合物的lcms分析表明完全转化至所需产物。反应物以冰水淬灭后,以0.5n盐酸酸化至ph=2~3,并以乙酸乙酯洗涤(10ml×3)。水层以浓碳酸钠水溶液碱化至ph=10~11,并以dcm萃取(20ml×3)。合并有机层经卤水洗涤、硫酸钠干燥及过滤后,浓缩至干态。残余物经硅胶纯化(石油醚:乙酸乙酯=0:60),获得米白色固体的n-(2,5-二氯嘧啶-4-基)-4-甲基烟酰胺(0.41g,收率:39.42%)。
831、lc-ms m/z:283.01(m+1)+。
832、步骤3:n-(5-氯-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)-4-甲基烟酰胺
833、
834、将溶于异丙醇(1.5ml)中的n-(2,5-二氯嘧啶-4-基)-4-甲基烟酰胺化合物(50mg,0.177mmol,1当量)及4-(4-甲基哌嗪-1-基)苯胺(37.16mg,0.194mmol,1.2当量)在100℃下搅拌1小时。lcms分析表明检测到所需物质且反应完成。冷却至室温后,将混合物过滤,然后小心倒入10ml的水和二氯甲烷中。通过加入盐酸使ph=5~6后,分离水相。水相以氢氧化钠水溶液碱化至ph=7~8,并以二氯甲烷萃取。有机层浓缩后,经制备型tlc纯化,获得淡黄色固体的实施例66。
835、lc-ms m/z:438.1(m+1)+;
836、1h nmr(400mhz,d6-dmso):δ9.63(s,1h),8.61(s,1h),8.53(d,j=5.0hz,1h),8.46(s,1h),7.51(d,j=8.7hz,2h),7.36(d,j=5.3hz,1h),6.86(d,j=8.5hz,2h),3.12(s,3h),2.80(s,3h),2.43(s,4h),1.20(s,4h)。
837、实施例67:
838、n-(5-氰基-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)-2-氟-6-甲氧基苯甲酰胺
839、
840、路线图
841、
842、步骤1:n-(2-氯-5-氰基嘧啶-4-基)-2-氟-6-甲氧基苯甲酰胺
843、
844、0℃下,在混于dmf(2ml)内的4-氨基-2-氯嘧啶-5-腈(181.68mg,1.18mmol,1当量)中,加入氢化钠(70.52mg,5.88mmol,1.5当量)。搅拌0.5小时后,在0℃下向反应物中逐滴加入溶于dcm(2ml)中的2-氟-6-甲氧基苯甲酰氯(221.00mg,1.18mmol,1当量)。反应室温搅拌0.5小时后,反应混合物的lcms分析表明完全转化至所需产物。反应物以水稀释后,混合物以1n盐酸水溶液酸化至ph=5~6,并以乙酸乙酯萃取。分离出的有机层中加入碳酸氢钠水溶液(20ml×3),并以水相萃取产物。水相以1n盐酸酸化,且以乙酸乙酯萃取。合并的有机萃取物经硫酸钠干燥、过滤及浓缩,获得米白色固体的n-(2-氯-5-氰基嘧啶-4-基)-2-氟-6-甲氧基苯甲酰胺(228.4mg,收率:63.4%)。
845、lc-ms m/z:307.0(m+1)+。
846、步骤2:n-(5-氰基-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)-2-氟-6-甲氧基苯甲酰胺
847、
848、将溶于1,4-二恶烷(1ml)中的n-(2-氯-5-氰基嘧啶-4-基)-2-氟-6-甲氧基苯甲酰胺(60mg,0.196mmol,1当量)、4-(4-甲基哌嗪-1-基)苯胺(37.42mg,0.196mmol,1当量)、碳酸铯(127.49mg,0.391mmol,2当量)、乙酸钯(4.39mg,0.020mmol,0.1当量)及xantphos(22.64mg,0.039mmol,0.2当量)通过微波反应器加热至100℃,并搅拌10分钟。反应物的lcms分析表明检测到所需物质且反应完成。将反应混合物过滤后,小心倒入25ml的水中,然后分离水相上的有机层。有机层经浓缩及硅胶纯化(甲醇/dcm=0:10%),获得黄色固体的实施例67(7.0mg,收率:5.8%)。
849、lc-ms m/z:462.2(m+1)+;
850、1h nmr(400mhz,d6-dmso):δ10.17(s,1h),8.71(s,1h),7.50(s,1h),7.49~7.42(m,2h),7.37~7.33(m,1h),7.00~6.86(m,2h),6.86~6.79(m,1h),3.78(s,3h),3.05(s,4h),2.44(s,4h),2.20(s,3h)。
851、实施例68:
852、n-(5-溴-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)-2-氟-6-甲氧基苯甲酰胺
853、
854、路线图3
855、
856、步骤1:n-(5-溴-2-氯嘧啶-4-基)-2-氟-6-甲氧基苯甲酰胺
857、
858、0℃下,在混于dmf(10ml)内的5-溴-2-氯嘧啶-4-胺(1.1g,5.29mmol,0.9当量),加入氢化钠(317mg,7.93mmol,1.35当量),并搅拌0.5小时。随后,在0℃下,向反应物中逐滴加入溶于dcm(10ml)中的2-氟-6-甲氧基苯甲酰氯(1.1g,5.88mmol,1当量),并将反应物室温搅拌2小时。反应混合物的lcms分析表明完全转化至所需产物。随后,加入水,并以dcm萃取(×3)产物。合并的有机萃取物经干燥(硫酸钠)、过滤及浓缩,残余物经硅胶纯化(石油醚/乙酸乙酯=0~100),获得淡黄色固体的n-(5-溴-2-氯嘧啶-4-基)-2-氟-6-甲氧基苯甲酰胺(436.7mg,收率:20.6%)。
859、lc-ms m/z:359.9(m+1)+。
860、步骤2:n-(5-溴-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)-2-氟-6-甲氧基苯甲酰胺
861、
862、将溶于1,4-二恶烷(1ml)中的n-(5-溴-2-氯嘧啶-4-基)-2-氟-6-甲氧基苯甲酰胺(100mg,0.28mmol,1当量)、4-(4-甲基哌嗪-1-基)苯胺(53mg,0.28mmol,1当量)、碳酸铯(181mg,0.55mmol,2当量)、乙酸钯(6mg,0.03mmol,0.1当量)及xantphos(32.1mg,0.06mmol,0.2当量)通过微波反应器加热至100℃,并搅拌20分钟。反应物的lcms分析表明检测到所需物质且反应完成。将反应混合物过滤后,小心倒入25ml的水中,然后分离水相上的有机层。有机层经浓缩及硅胶纯化(甲醇/dcm=0:10%),获得黄色固体的实施例68(4.2mg,收率:2.7%)。
863、lc-ms m/z:515.1(m+1)+;
864、1h nmr(400mhz,d6-dmso):δ10.57(s,1h),9.53(s,1h),8.47(s,1h),7.48~7.41(m,2h),7.40(s,1h),6.92
865、(d,j=8.5hz,1h),6.86(t,j=8.7hz,1h),6.81(d,j=8.8hz,2h),3.76(s,3h),3.03(s,4h),2.44(s,4h),2.20(s,3h)。
866、按照实施例53的方式,合成如下化合物:
867、
868、
869、
870、
871、
872、
873、按照实施例60的方法,合成如下化合物:
874、
875、
876、
877、
878、
879、
880、
881、
882、
883、按照实施例231的方法,合成实施例102。
884、
885、
886、实施例103:
887、5-氯-n-(3-氟-4-(4-甲基哌嗪-1-基)苯基)-4-(1-(氧杂环丁-3-基)-1h-苯并[d]咪唑-6-基)嘧啶-2-胺
888、
889、路线图
890、
891、步骤1:n-(5-氯-4-(1-(氧杂环丁-3-基)-1h-苯并[d]咪唑-6-基)嘧啶-2-基)-n-(3-氟-4-(4-甲基哌嗪-1-基)苯基)甲酰胺
892、
893、氮气气氛中,在溶于二恶烷(1.5ml)内的6-(2,5-二氯嘧啶-4-基)-1-(氧杂环丁-3-基)-1h-苯并[d]咪唑(100mg,311.37μmol,1当量)及n-(3-氟-4-(4-甲基哌嗪-1-基)苯基)甲酰胺(73.88mg,311.37μmol,1当量),加入xantphos(47.4mg,62.2μmol,0.2当量)、碳酸铯(202.9mg,622.74μmol,2当量)及乙酸钯(7.2mg,31.1μmol,0.1当量)。所得混合物在100℃下微波搅拌2小时,获得棕色悬浮液。lcms表明起始物质已完全消耗且检测到一个具有所需物质的主峰。反应混合物冷却至室温后,经过滤和浓缩,获得棕色油状物的n-(5-氯-4-(1-(氧杂环丁-3-基)-1h-苯并[d]咪唑-6-基)嘧啶-2-基)-n-(3-氟-4-(4-甲基哌嗪-1-基)苯基)甲酰胺。
894、lc-ms m/z:523(m+1)+。
895、步骤2:5-氯-n-(3-氟-4-(4-甲基哌嗪-1-基)苯基)-4-(1-(氧杂环丁-3-基)-1h-苯并[d]咪唑-6-基)嘧啶-2-胺
896、
897、在溶于甲醇(1ml)内的n-(5-氯-4-(1-(氧杂环丁-3-基)-1h-苯并[d]咪唑-6-基)嘧啶-2-基)-n-(3-氟-4-(4-甲基哌嗪-1-基)苯基)甲酰胺(311.3μmol,1当量)中,加入氢氧化钠(0.5ml),并在60℃下搅拌3小时。lcms表明起始物质已大部分消耗且检测到一个峰。反应物以水稀释,并以乙酸乙酯萃取。残余物经c18色谱纯化(0%~100%的乙腈水溶液,combiflash nextgen 300),获得黄色固体的实施例103化合物(43mg,28%,甲酸盐)。
898、lc-ms m/z:494.18(m+1)+;
899、1h nmr(400mhz,甲醇-d4)δ9.14(s,1h),8.69(t,j=1.1hz,1h),8.58(s,1h),8.12(dd,j=8.6,1.6hz,1h),7.97(d,j=8.6hz,1h),7.84(dd,j=15.0,2.5hz,1h),7.39(ddd,j=8.7,2.6,1.1hz,1h),7.07(t,j=9.2hz,1h),5.92(tt,j=7.5,5.3hz,1h),5.33(t,j=7.6hz,2h),5.20(dd,j=7.8,5.3hz,2h),3.61(d,j=12.2hz,2h),3.54(d,j=13.2hz,2h),3.37(d,j=2.8hz,2h),3.20~3.05(m,2h),3.00(s,3h)。
900、实施例104:
901、5-氯-n-(2-氰基-6-氟苯基)-2-((6-甲基-5,6,7,8-四氢-1,6-萘啶-3-基)氨基)嘧啶-4-羧酰胺
902、
903、路线图
904、
905、步骤1:n-(6-甲基-5,6,7,8-四氢-1,6-萘啶-3-基)甲酰胺
906、
907、将溶于甲酸(15ml)中的6-甲基-5,6,7,8-四氢-1,6-萘啶-3-胺(3.00g,18.38mmol,1.0当量)在110℃下搅拌3小时,获得棕色溶液。反应混合物的lcms分析表明完全转化至所需产物。混合物以碳酸氢钠调节至ph=6~7,并减压浓缩至干态,获得灰色固体的n-(6-甲基-5,6,7,8-四氢-1,6-萘啶-3-基)甲酰胺化合物(3.51g,收率:100%)。
908、lc-ms m/z:192.1(m+1)+。
909、步骤2:5-氯-2-(n-(6-甲基-5,6,7,8-四氢-1,6-萘啶-3-基)甲酰胺基)嘧啶-4-羧酸甲酯
910、
911、在溶于thf(15ml)和dmf(5ml)内的n-(6-甲基-5,6,7,8-四氢-1,6-萘啶-3-基)甲酰胺(520mg,2.72mmol,1.0当量)中,加入氢化钠(163.14mg,4.08mmol,1.5当量),并在50℃下搅拌40分钟。随后,分批加入5-氯-2-(甲磺酰基)嘧啶-4-羧酸甲酯(1.02g,4.08μmol,1.5当量)。反应物在26℃下搅拌过夜,获得棕色悬浮液。lcms表明主峰为所需物质且检测到所需物质。反应物以thf稀释,并以硅藻土垫过滤。有机相减压浓缩至干态后,粗产物经快速柱纯化(二氧化硅,以溶有0~10%dcm的甲醇洗脱,combiflash nextgen 300),获得黄色固体的5-氯-2-(n-(6-甲基-5,6,7,8-四氢-1,6-萘啶-3-基)甲酰胺基)嘧啶-4-羧酸甲酯化合物(361.7mg,收率:37%)。
912、lc-ms m/z:362(m+1)+。
913、步骤3:5-氯-2-((6-甲基-5,6,7,8-四氢-1,6-萘啶-3-基)氨基)嘧啶-4-羧酸
914、
915、将加入二恶烷(5ml)中的5-氯-2-(n-(6-甲基-5,6,7,8-四氢-1,6-萘啶-3-基)甲酰胺基)嘧啶-4-羧酸甲酯(361.70mg,0.99mmol,1.0当量)的4m盐酸溶液在26℃下搅拌过夜,获得黄色悬浮液。lcms表明反应完成。混合物减压浓缩至干态后,加入10%氢氧化钠水溶液(5ml),并在26℃下搅拌10分钟,获得黄色悬浮液。混合物减压浓缩至干态,并以柠檬酸调节至ph=5~6。混合物浓缩至干态后,获得黄色固体的5-氯-2-(n-(6-甲基-5,6,7,8-四氢-1,6-萘啶-3-基)甲酰胺基)嘧啶-4-羧酸甲酯化合物(476.7mg,收率:149%)。
916、lc-ms m/z:320(m+1)+。
917、步骤4:5-氯-n-(2-氰基-6-氟苯基)-2-((6-甲基-5,6,7,8-四氢-1,6-萘啶-3-基)氨基)嘧啶-4-羧酰胺
918、
919、在溶于吡啶(3ml)内的5-氯-2-(n-(6-甲基-5,6,7,8-四氢-1,6-萘啶-3-基)甲酰胺基)嘧啶-4-羧酸甲酯(150mg,469.12μmol,1.0当量)中,加入2-氨基-3-氟苯腈(95.79mg,703.68μmol,1.5当量),并分批加入三氯氧磷(0.15ml)。反应物在26℃下搅拌15分钟,获得深棕色溶液。lcms表明其中一个峰为所需物质且起始物质几乎消耗殆尽。混合物倒入冰水中,并以碳酸氢钠洗涤且以dcm萃取(3×)。有机相减压浓缩至干态,粗产物经快速柱纯化(c18,出峰时间=40%的乙腈水溶液,combiflash nextgen 300),获得黄色固体的实施例104化合物(35.4mg,收率:17.2%)。
920、lc-ms m/z:438.12(m+1)+;
921、1h nmr(400mhz,d6-meod):δ8.72(s,1h),8.64(s,1h),8.53(s,1h),7.82~7.55(m,3h),4.59(s,2h),3.70(d,j=32.4hz,2h),3.28(t,j=5.6hz,2h),3.12(s,3h)。
922、
923、
924、
925、
926、
927、
928、实施例118:
929、5-氯-n-(2-氯-6-氰基苯基)-2-((3-甲氧基-4-(4-(4-甲基哌嗪-1-基)哌啶-1-基)苯基)氨基)嘧啶-4-羧酰胺
930、
931、路线图
932、
933、步骤1:n-(3-甲氧基-4-(4-(4-甲基哌嗪-1-基)哌啶-1-基)苯基)甲酰胺
934、
935、将3-甲氧基-4-(4-(4-甲基哌嗪-1-基)哌啶-1-基)苯胺(2.80g,9.20mmol,1.00当量)与甲酸(30ml)的混合物回流搅拌4小时,获得黑色溶液。反应混合物的lc-ms分析表明完全转化至所需产物。反应混合物在真空中浓缩至干态后,加入饱和碳酸氢钠溶液,调至ph=8~9。冻干后,加入甲醇(50ml),并经过滤和干燥,获得紫色固体的n-(3-甲氧基-4-(4-(4-甲基哌嗪-1-基)哌啶-1-基)苯基)甲酰胺化合物(3.0g,收率100%)。
936、lc-ms m/z:333.5(m+1)+。
937、步骤2:5-氯-2-(n-(3-甲氧基-4-(4-(4-甲基哌嗪-1-基)哌啶-1-基)苯基)甲酰胺基)嘧啶-4-羧酸甲酯
938、
939、在混于thf(40ml)内的n-(3-甲氧基-4-(4-(4-甲基哌嗪-1-基)哌啶-1-基)苯基)甲酰胺化合物(1.00g,3.01mmol,1.0当量)中,加入氢化钠(0.18g,4.50mmol,1.50当量),并在50℃下搅拌0.5小时,获得灰色悬浮液,随后加入5-氯-2-(甲磺酰基)嘧啶-4-羧酸酯(1.13g,4.50mmol,1.50当量)。混合物在氮气气氛中50℃搅拌5小时,获得棕色悬浮液。lcms分析显示大部分反应混合物转化为所需产物。混合物中加入乙酸(1ml)后,进行过滤。滤饼以thf(20ml)洗涤后,减压浓缩。残余物经快速柱纯化(二氧化硅,以溶有20%~30%甲醇的dcm洗脱,combiflash nextgen 300),获得淡黄色固体的5-氯-2-(n-(3-甲氧基-4-(4-(4-甲基哌嗪-1-基)哌啶-1-基)苯基)甲酰胺基)嘧啶-4-羧酸甲酯化合物(1.10g,收率:66%)。
940、lc-ms m/z:504(m+1)+。
941、步骤3:5-氯-2-((3-甲氧基-4-(4-(4-甲基哌嗪-1-基)哌啶-1-基)苯基)氨基)嘧啶-4-羧酸
942、
943、在混于甲醇(10ml)内的5-氯-2-(n-(3-甲氧基-4-(4-(4-甲基哌嗪-1-基)哌啶-1-基)苯基)甲酰胺基)嘧啶-4-羧酸甲酯(1.10g,2.63mmol,1.00当量)中,加入氢氧化钠(0.32g,7.90mmol,3.00当量)和水(5ml),搅拌2小时后,获得棕色溶液。反应混合物的lcms分析表明完全转化至所需产物。随后,通过减压浓缩,去除多余的甲醇。残余物加入柠檬酸(2.5g)后,进行真空干燥,并经快速柱纯化(c18,0~100% a(水+0.5%甲酸),40%~60%b(偶氮二环己腈)(accn)+0.5%甲酸),combiflash nextgen 300),获得棕色固体的5-氯-2-((3-甲氧基-4-(4-(4-甲基哌嗪-1-基)哌啶-1-基)苯基)氨基)嘧啶-4-羧酸化合物(1.2g,收率:99%)。
944、lc-ms m/z:462.0(m+1)+。
945、步骤4:5-氯-n-(2-氯-6-氰基苯基)-2-((3-甲氧基-4-(4-(4-甲基哌嗪-1-基)哌啶-1-基)苯基)氨基)嘧啶-4-羧酰胺
946、
947、在混于吡啶(5ml)内的5-氯-2-((3-甲氧基-4-(4-(4-甲基哌嗪-1-基)哌啶-1-基)苯基)氨基)嘧啶-4-羧酸(100mg,0.22mmol,1.0当量)及2-氨基-3-甲氧基苯腈(67mg,0.44mmol,2.00当量)中,加入三氯氧磷(101mg,0.66mmol,3.00当量)。混合物在25℃下搅拌0.5小时,获得黑色溶液。反应混合物的lcms分析表明反应完成。混合物倒入饱和碳酸氢钠溶液(50ml)中,并以乙酸乙酯(20ml×2)萃取。有机层经硫酸钠干燥后,在真空中浓缩至干态。残余物经快速柱纯化(c18,0~100% a(水+0.5%甲酸),50%~60%b(accn+0.5%甲酸),combiflash nextgen 300),获得棕色固体的实施例118化合物(11mg,收率:8%,三氟乙酸盐)。
948、lc-ms m/z:596.5(m+1)+;
949、1h nmr(400mhz,meod)δ8.74(s,1h),7.93(dd,j=8.2,1.3hz,1h),7.87(dd,j=5.5,2.2hz,2h),7.65~7.46(m,3h),4.08(s,3h),3.80~3.61(m,11h),3.15(m,2h),2.96(s,2h),2.30(s,2h),2.23~2.12(m,1h)。
950、
951、
952、
953、
954、
955、实施例128:
956、5-氯-n-(2-氯-6-氟苯基)-2-((4-(4-(4-甲基哌嗪-1-基)哌啶-1-基)苯基)氨基)嘧啶-4-羧酰胺
957、
958、路线图
959、
960、步骤1:n-(4-(4-(4-甲基哌嗪-1-基)哌啶-1-基)苯基)甲酰胺
961、
962、将溶于甲酸(8ml)中的4-(4-(4-甲基哌嗪-1-基)哌啶-1-基)苯胺(3g,10.93mmol,1.0当量)在100℃下搅拌1小时,获得紫色溶液。lcms表明反应完成。混合物以碳酸钠调至ph=6~7,并减压浓缩至干态,获得紫色固体的n-(4-(4-(4-甲基哌嗪-1-基)哌啶-1-基)苯基)甲酰胺化合物(3.77g,收率:114%)。
963、lc-ms m/z:303.21(m+1)+。
964、步骤2:5-氯-2-((4-(4-(4-甲基哌嗪-1-基)哌啶-1-基)苯基)氨基)嘧啶-4-羧酸
965、
966、在溶于dmf(20ml)内的n-(4-(4-(4-甲基哌嗪-1-基)哌啶-1-基)苯基)甲酰胺(2g,6.61mmol,1.0当量)中,加入氢化钠(0.4g,9.92mmol,1.5当量),并在50℃下搅拌40分钟,随后分批加入5-氯-2-(甲磺酰基)嘧啶-4-羧酸甲酯(2.49g,9.92mmol,1.5当量)。反应物在20℃下搅拌过夜,获得棕色悬浮液。lcms表明其中一个主峰为所需物质且起始物质几乎消耗殆尽。反应物经thf洗涤及硅藻土垫过滤后,滤过物减压浓缩至干态。混合物中加入甲醇(5ml)及10%氢氧化钠水溶液(5ml),并在20℃下搅拌2小时,获得棕色悬浮液。lcms表明主峰为所需物质且起始物质几乎消耗殆尽。反应物减压浓缩至干态后,以柠檬酸调节至ph=5~6。混合物以水洗涤且以硅藻土垫过滤后,滤饼进行浓缩,获得奶油色固体的5-氯-2-((4-(4-(4-甲基哌嗪-1-基)哌啶-1-基)苯基)氨基)嘧啶-4-羧酸化合物(1.238g,收率:40%)。
967、lc-ms m/z:431.19(m+1)+。
968、步骤3:5-氯-n-(2-氯-6-氟苯基)-2-((4-(4-(4-甲基哌嗪-1-基)哌啶-1-基)苯基)氨基)嘧啶-4-羧酰胺
969、
970、在溶于吡啶(2ml)内的5-氯-2-((4-(4-(4-甲基哌嗪-1-基)哌啶-1-基)苯基)氨基)嘧啶-4-羧酸(100mg,232.05μmol,1.0当量)中,加入2-氯-6-氟苯胺(101.33mg,696.16μmol,3.0当量),并分批加入三氯氧磷(0.06ml)。反应物在21℃下搅拌15分钟,获得深棕色溶液。lcms表明其中一个主峰为所需物质且起始物质几乎消耗殆尽。混合物倒入冰水中后,以碳酸氢钠洗涤,并以dcm萃取(3×)。有机相减压浓缩至干态后,粗产物经快速柱纯化(c18,出峰时间=30%的乙腈水溶液,combiflash nextgen 300),获得黄色固体的实施例128化合物(80.9mg,收率:62.4%)。
971、lc-ms m/z:558.19(m+1)+。
972、1h nmr(400mhz,d6-meod):δ8.55(s,1h),7.61(d,j=8.9hz,2h),7.45~7.38(m,2h),7.27(m,j=9.6,6.2,3.6hz,1h),7.01(d,j=9.0hz,2h),3.72(d,j=11.8hz,2h),3.12~2.58(m,14h),2.05(d,j=11.6hz,2h),1.73(d,j=9.9hz,2h)。
973、
974、
975、
976、
977、实施例140:
978、5-氯-n-(2-氰基-6-甲氧基苯基)-2-((4-(4-异丙基哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺
979、
980、路线图
981、
982、步骤1:4-(4-甲酰胺基苯基)哌嗪-1-羧酸叔丁酯
983、
984、将溶于甲酸乙酯(55ml)中的4-(4-氨基苯基)哌嗪-1-羧酸叔丁酯(10.2g,36.77mmol,1.0当量)在70℃下搅拌2天,获得棕色溶液。lcms表明反应完成。混合物减压浓缩至干态,获得灰色固体的4-(4-甲酰胺基苯基)哌嗪-1-羧酸叔丁酯化合物(11.17g,收率:99.5%)。
985、lc-ms m/z:306.17(m+1)+。
986、步骤2:2-((4-(4-(叔丁氧基羰基)哌嗪-1-基)苯基)氨基)-5-氯嘧啶-4-羧酸甲酯
987、
988、在溶于thf(5ml)内的4-(4-甲酰胺基苯基)哌嗪-1-羧酸叔丁酯(174.30mg,570.77μmol,1.0当量)中,加入氢化钠(34.24mg,856.15μmol,1.5当量)。50℃下搅拌30分钟后,在26℃下分批加入5-氯-2-(甲磺酰基)嘧啶-4-羧酸甲酯(214.60mg,856.15μmol,1.5当量)。反应物在26℃下搅拌过夜,获得黄色悬浮液。lcms表明其中一个主峰为所需物质且起始物质仍有残留。反应物以thf稀释,并以硅藻土垫过滤。有机相减压浓缩至干态后,粗产物经快速柱纯化(二氧化硅,以溶有0~60%乙酸乙酯的石油醚洗脱,combiflash nextgen 300),获得棕色油状液体的2-((4-(4-(叔丁氧基羰基)哌嗪-1-基)苯基)氨基)-5-氯嘧啶-4-羧酸甲酯化合物(232.8mg,收率:91%)。
989、lc-ms m/z:448.17(m+1)+。
990、步骤3:2-((4-(4-(叔丁氧基羰基)哌嗪-1-基)苯基)氨基)-5-氯嘧啶-4-羧酸
991、
992、将溶于甲醇(3ml)和10%氢氧化钠水溶液(3ml)中的2-((4-(4-(叔丁氧基羰基)哌嗪-1-基)苯基)氨基)-5-氯嘧啶-4-羧酸甲酯(232.8mg,519.74μmol,1.0当量)26℃下搅拌1小时,获得黄色悬浮液。lcms表明主峰为所需物质且起始物质几乎消耗殆尽。混合物减压浓缩至干态后,以柠檬酸调节至ph=5~6。混合物浓缩至干态,获得淡黄色固体的2-((4-(4-(叔丁氧基羰基)哌嗪-1-基)苯基)氨基)-5-氯嘧啶-4-羧酸(473.6mg,收率:210%)化合物。
993、lc-ms m/z:434.15(m+1)+。
994、步骤4:5-氯-n-(2-氰基-6-甲氧基苯基)-2-((4-(哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺
995、
996、在溶于吡啶(2ml)内的2-((4-(4-(叔丁氧基羰基)哌嗪-1-基)苯基)氨基)-5-氯嘧啶-4-羧酸(100mg,230.47μmol,1.0当量)中,加入2-氨基-3-甲氧基苯腈(68.30mg,460.94μmol,2.0当量),并分批加入三氯氧磷(0.2ml)。反应物在26℃下搅拌3小时,获得深棕色溶液。lcms表明其中一个峰为所需物质且起始物质几乎消耗殆尽。混合物减压浓缩至干态后,粗产物经快速柱纯化(二氧化硅,以溶有0~60%乙酸乙酯的石油醚洗脱,combiflashnextgen 300),获得棕色油状液体的5-氯-n-(2-氰基-6-甲氧基苯基)-2-((4-(哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺化合物。
997、lc-ms m/z:564.2(m+1)+。
998、步骤5:5-氯-n-(2-氰基-6-甲氧基苯基)-2-((4-(哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺
999、
1000、将加入二恶烷(1ml)中的5-氯-n-(2-氰基-6-甲氧基苯基)-2-((4-(哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺的4m盐酸溶液在26℃下搅拌2小时,获得棕色溶液。lcms表明其中一个峰为所需物质且起始物质几乎消耗殆尽。混合物减压浓缩至干态后,粗产物经快速柱纯化(c18,出峰时间=40%的乙腈水溶液,combiflash nextgen 300),获得黄色固体的5-氯-n-(2-氰基-6-甲氧基苯基)-2-((4-(哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺化合物(4.6mg)。
1001、lc-ms m/z:464.15(m+1)+。
1002、步骤6:5-氯-n-(2-氰基-6-甲氧基苯基)-2-((4-(4-异丙基哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺
1003、
1004、在溶于dcm(2ml)内的5-氯-n-(2-氰基-6-甲氧基苯基)-2-((4-(哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺(100mg,215.55μmol,1.0当量)中,加入丙酮(0.06ml)和三乙酰氧基硼氢化钠(stab)(68.53mg,323.33μmol,1.0当量)。反应物在26℃下搅拌过夜,获得黄色悬浮液。lcms表明主峰为所需物质且起始物质几乎消耗殆尽。混合物减压浓缩至干态后,粗产物经快速柱纯化(c18,出峰时间=50%的乙腈水溶液,combiflash nextgen 300),获得黄色固体的实施例140化合物(44.6mg,收率:41%)。
1005、lc-ms m/z:506.20(m+1)+。
1006、1h nmr(400mhz,d6-meod):δ8.57(s,1h),7.69(d,j=8.9hz,2h),7.49(dt,j=15.1,7.7hz,2h),7.41(d,j=7.4hz,1h),7.05(d,j=9.0hz,2h),3.95(s,3h),3.84(d,j=13.6hz,2h),3.62(dd,j=13.3,6.8hz,3h),3.35(s,2h),3.04(t,j=12.1hz,2h),1.44(d,j=6.7hz,6h)。
1007、
1008、
1009、
1010、
1011、
1012、
1013、
1014、
1015、
1016、实施例164:5-氯-n-(2-氰基-6-甲氧基苯基)-2-((3-甲基-4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺
1017、
1018、路线图
1019、
1020、步骤1:n-(3-甲基-4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯基)甲酰胺
1021、
1022、将溶于甲酸(12ml)中的3-甲基-4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯胺(4.41g,15.29mmol,1.0当量)在100℃下搅拌1小时,获得紫色溶液。lcms表明反应完成。混合物以碳酸钠调至ph=6~7后,减压浓缩至干态,获得棕色固体的n-(3-甲基-4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯基)甲酰胺化合物(6.78g,收率:140%)。
1023、lc-ms m/z:317.23(m+1)+。
1024、步骤2:5-氯-2-((3-甲基-4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯基)氨基)嘧啶-4-羧酸
1025、
1026、在溶于dmf(10ml)内的n-(3-甲基-4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯基)甲酰胺(1g,3.16mmol,1.0当量)中,加入氢化钠(0.19g,4.74mmol,1.5当量),并在50℃下搅拌40分钟,随后分批加入5-氯-2-(甲磺酰基)嘧啶-4-羧酸甲酯(1.19g,4.74mmol,1.5当量)。反应物在22℃下搅拌过夜,获得棕色悬浮液。lcms表明其中一个主峰为所需物质且起始物质几乎消耗殆尽。反应物以thf洗涤,并以硅藻土垫过滤。滤过物减压浓缩至干态后,在混合物内加入甲醇(3ml)以及10%的氢氧化钠水溶液(3ml),并在22℃下搅拌2小时,获得棕色悬浮液。lcms表明主峰为所需物质且起始物质几乎消耗殆尽。反应物减压浓缩至干态后,以柠檬酸调节至ph=5~6。混合物以水洗涤,并进行过滤。滤饼干燥后,获得奶油色固体的5-氯-2-((3-甲基-4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯基)氨基)嘧啶-4-羧酸化合物(978.3mg,收率69.4%)。
1027、lc-ms m/z:445.20(m+1)+。
1028、步骤3:5-氯-n-(2-氰基-6-甲氧基苯基)-2-((3-甲基-4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺
1029、
1030、在溶于吡啶(2ml)内的5-氯-2-((3-甲基-4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯基)氨基)嘧啶-4-羧酸(80mg,179.79μmol,1.0当量)中,加入2-氨基-3-甲氧基苯腈(79.92mg,539.37μmol,3.0当量),并分批加入三氯氧磷(0.06ml)。反应物在22℃下搅拌10分钟,获得深棕色溶液。lcms表明未检测到所需物质且起始物质几乎消耗殆尽。混合物倒入冰水中,并以碳酸氢钠洗涤,且以dcm萃取(3×)。有机相减压浓缩至干态后,粗产物经快速柱纯化(c18,出峰时间=30%的乙腈水溶液,combiflash nextgen 300),获得黄色固体的实施例164化合物
1031、(42.32mg,收率41%)。
1032、lc-ms m/z:575.26(m+1)+;
1033、1h nmr(400mhz,d6-meod):δ8.57(s,1h),7.59(d,j=7.6hz,1h),7.49(p,j=8.3hz,3h),7.40(d,j=7.3hz,1h),7.06(t,j=8.5hz,1h),3.97(d,j=14.5hz,3h),3.50(s,2h),2.96(d,j=28.1hz,10h),2.79(s,4h),2.31(d,j=10.3hz,3h),2.21(s,2h),1.91(s,2h)。
1034、
1035、
1036、实施例167:
1037、5-氯-n-(2,6-二氯苯基)-2-((3-甲氧基-4-(4-(4-甲基哌嗪-1-基)哌啶-1-基)苯基)氨基)嘧啶-4-羧酰胺
1038、
1039、路线图
1040、
1041、步骤1:5-氯-n-(2,6-二氯苯基)-2-(甲硫基)嘧啶-4-羧酰胺
1042、
1043、在混于吡啶内的2,6-二氯苯胺(10g,48.87mmol,1.0当量)及5-氯-2-(甲硫基)嘧啶-4-羧酸(7.92g,48.87mmol,1当量)中,加入三氯氧磷(13.62ml,146.61mmol,3当量)。混合物以氮气脱气3次,并在氮气气氛中室温搅拌2小时。反应混合物的lcms分析表明完全转化至所需产物。反应混合物倒入水(100ml)中,并以二氯甲烷(100ml×3)过滤。减压浓缩后,获得粗产物。粗产物经硅胶纯化(石油醚:乙酸乙酯=100:0~60:40),获得橙色固体的5-氯-n-(2,6-二氯苯基)-2-(甲硫基)嘧啶-4-羧酰胺(8.9g,52.24%)。
1044、lc-ms m/z:348.0(m+1)+。
1045、步骤2:5-氯-n-(2,6-二氯苯基)-2-(甲磺酰基)嘧啶-4-羧酰胺
1046、
1047、在溶于二氯甲烷(100ml)内的5-氯-n-(2,6-二氯苯基)-2-(甲硫基)嘧啶-4-羧酰胺(8.9g,25.53mmol,1当量)中,分数次加入间氯过氧苯甲酸(m-cpba)(18.14g,89.35mmol,3.5当量)。混合物室温搅拌1小时后,反应混合物的lcms分析表明完全转化至所需产物。反应混合物经过滤和dcm洗涤,有机相在真空中浓缩至干态,并经硅胶纯化(石油醚:二氯甲烷,100:0~0:10),获得淡黄色固体的5-氯-n-(2,6-二氯苯基)-2-(甲磺酰基)嘧啶-4-羧酰胺(8.48g,收率:87.27%)。
1048、lc-ms m/z:380.0(m+1)+。
1049、步骤3:5-氯-n-(2,6-二氯苯基)-2-((3-甲氧基-4-(4-(4-甲基哌嗪-1-基)哌啶-1-基)苯基)氨基)嘧啶-4-羧酰胺
1050、
1051、室温下,在溶于dmf(1ml)内的n-(3-甲氧基-4-(4-(4-甲基哌嗪-1-基)哌啶-1-基)苯基)甲酰胺(52.41mg,0.158mmol,1.2当量)中,加入氢化钠(9.46mg,0.236mmol,1.8当量)。随后,混合物在60℃下搅拌0.5小时,获得棕色溶液。搅拌0.5小时后,加入5-氯-n-(2-甲氧基-6-(三氟甲基)苯基)-2-(甲磺酰基)嘧啶-4-羧酰胺(50mg,0.131mmol,1当量)。混合物在氮气气氛中室温搅拌3小时后,反应混合物的lcms分析表明完全转化至所需产物。混合物过滤后,滤饼以dcm洗涤,并经减压浓缩,去除多余的溶剂。残余物经c18纯化(水:乙腈=90:10~95:05,水/乙腈中溶有0.1%甲酸),获得黄色固体的实施例167化合物(55.5mg,收率:69.2%)。
1052、lc-ms m/z:604.17(m+1)+;
1053、1h nmr(400mhz,meod):δ8.73(s,1h),7.94(d,j=2.1hz,1h),7.64(d,j=9.0hz,1h),7.60~7.54(m,3h),7.41(dd,j=8.6,7.7hz,1h),4.08(s,3h),3.95~3.44(m,13h),3.03(d,j=10.4hz,3h),2.54(s,2h),2.44(s,2h)。
1054、
1055、
1056、
1057、
1058、
1059、实施例178:2-(4-(4-((5-氯-4-((2,6-二氯苯基)氨基甲酰基)嘧啶-2-基)氨基)苯基)哌嗪-1-基)乙酸
1060、
1061、路线图
1062、
1063、步骤1:5-氯-n-(2,6-二氯苯基)-2-(甲硫基)嘧啶-4-羧酰胺
1064、
1065、在混于吡啶内的2,6-二氯苯胺(10g,48.87mmol,1.0当量)及5-氯-2-(甲硫基)嘧啶-4-羧酸(7.92g,48.87mmol,1当量)中,加入三氯氧磷(13.62ml,146.61mmol,3当量)。混合物以氮气脱气3次,并在氮气气氛中室温搅拌2小时。反应混合物的lcms分析表明完全转化至所需产物。反应混合物倒入水中(100ml)后,经二氯甲烷(100ml×3)过滤及减压浓缩,获得粗产物。粗产物经硅胶纯化(石油醚:乙酸乙酯=100:0~60:40),获得橙色固体的5-氯-n-(2,6-二氯苯基)-2-(甲硫基)嘧啶-4-羧酰胺(8.9g,52.24%)。
1066、lc-ms m/z:348.0(m+1)+。
1067、步骤2:5-氯-n-(2,6-二氯苯基)-2-(甲磺酰基)嘧啶-4-羧酰胺
1068、
1069、在溶于二氯甲烷(100ml)内的5-氯-n-(2,6-二氯苯基)-2-(甲硫基)嘧啶-4-羧酰胺(8.9g,25.53mmol,1当量)中,分数次加入m-cpba(18.14g,89.35mmol,3.5当量)。混合物室温搅拌1小时后,反应混合物的lcms分析表明完全转化至所需产物。反应混合物经过滤和dcm洗涤,有机相在真空中浓缩至干态,并经硅胶纯化(石油醚:二氯甲烷,100:0~0:10),获得淡黄色固体的5-氯-n-(2,6-二氯苯基)-2-(甲磺酰基)嘧啶-4-羧酰胺(8.48g,收率:87.27%)。
1070、lc-ms m/z:380.0(m+1)+。
1071、步骤3:4-(4-(n-(5-氯-4-((2,6-二氯苯基)氨基甲酰基)嘧啶-2-基)甲酰胺基)苯基)哌嗪-1-羧酸叔丁酯
1072、
1073、室温下,在溶于dmf(10ml)内的4-(4-甲酰胺基苯基)哌嗪-1-羧酸叔丁酯(0.883g,2.89mmol,1.1当量)中,加入氢化钠(0.158g,3.94mmol,1.5当量)。随后,混合物在60℃下搅拌0.5小时,获得深绿色溶液。搅拌0.5小时后,加入5-氯-n-(2,6-二氯苯基)-2-(甲磺酰基)嘧啶-4-羧酰胺(1g,2.63mmol,1当量)。混合物在氮气气氛中室温搅拌3小时后,反应混合物的lcms分析表明完全转化至所需产物。混合物过滤后,滤饼经乙腈洗涤,并经减压浓缩,去除多余的溶剂。残余物经c18纯化(水:乙腈=90:10~95:05,水/乙腈中溶有0.1%的甲酸),获得黄色固体的4-(4-(n-(5-氯-4-((2,6-二氯苯基)氨基甲酰基)嘧啶-2-基)甲酰胺基)苯基)哌嗪-1-羧酸叔丁酯(980mg,收率:61.6%)。
1074、lc-ms m/z:605.12(m+1)+。
1075、步骤4:5-氯-n-(2,6-二氯苯基)-2-((4-(哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺
1076、
1077、在溶于dcm(4ml)内的4-(4-(n-(5-氯-4-((2,6-二氯苯基)氨基甲酰基)嘧啶-2-基)甲酰胺基)苯基)哌嗪-1-羧酸叔丁酯(540mg,0.891mmol,1当量)中,加入99%的tfa(2ml),随后将混合物室温搅拌0.5小时,获得橙色溶液。反应混合物的lcms分析表明完全转化至所需产物。反应物中加入碳酸钠水溶液,并以dcm萃取。有机相浓缩后,获得橙色固体的5-氯-n-(2,6-二氯苯基)-2-((4-(哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺(370mg,收率:86.9%)。
1078、lc-ms m/z:477.07(m+1)+。
1079、步骤5:5-氯-n-(2,6-二氯苯基)-2-((4-(哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺
1080、
1081、在溶于dmf(1ml)内的5-氯-n-(2,6-二氯苯基)-2-((4-(哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺(50mg,0.105mmol,1.1当量)中,加入2-溴乙酸(13.22mg,0.095mmol,1当量)及碳酸钾(26.3mg,0.190mmol,2当量)。反应物在微波反应器内120℃搅拌2小时获得橙色悬浮液。反应混合物的lcms分析表明完全转化至所需产物。混合物过滤后,滤饼以乙腈洗涤后减压浓缩,去除多余的溶剂。残余物经c18纯化(水:乙腈=90:10~95:05,水/乙腈中溶有0.1%的甲酸),获得黄色固体的实施例178化合物(15.9mg,收率:31.2%)。
1082、lc-ms m/z:533.07(m-1)-;
1083、1h nmr(400mhz,meod):δ8.57(s,1h),7.69(d,j=9.0hz,2h),7.57(s,1h),7.55(s,1h),7.39(dd,j=8.6,7.6hz,1h),7.03(d,j=9.1hz,2h),3.72(s,2h),3.51(d,j=3.4hz,4h),3.44(s,4h)。
1084、
1085、
1086、
1087、实施例184:5-氯-n-(2-甲氧基-6-(三氟甲基)苯基)-2-((4-(4-甲基哌嗪-1-基)-3-(三氟甲基)苯基)氨基)嘧啶-4-羧酰胺
1088、
1089、路线图
1090、
1091、步骤1:5-氯-n-(2-甲氧基-6-(三氟甲基)苯基)-2-(甲硫基)嘧啶-4-羧酰胺
1092、
1093、在混于吡啶内的2-甲氧基-6-(三氟甲基)苯胺(1.5g,7.85mmol,1.0当量)及5-氯-2-(甲硫基)嘧啶-4-羧酸(1.77g,8.63mmol,1.1当量)中,加入三氯氧磷(2.55ml,27.46mmol,3.5当量)。混合物以氮气脱气3次,并在氮气气氛中室温搅拌2小时。反应混合物的lcms分析表明完全转化至所需产物。反应混合物倒入水中(50ml),并经二氯甲烷过滤(50ml×3)和减压浓缩,获得粗产物。粗产物经硅胶纯化(石油醚:乙酸乙酯=100:0~60:40),获得黄色固体的y5-氯-n-(2-甲氧基-6-(三氟甲基)苯基)-2-(甲硫基)嘧啶-4-羧酰胺(1.66g,56.1%)。
1094、lc-ms m/z:378.02(m+1)+。
1095、步骤2:5-氯-n-(2-甲氧基-6-(三氟甲基)苯基)-2-(甲磺酰基)嘧啶-4-羧酰胺
1096、
1097、在溶于二氯甲烷(20ml)内的y5-氯-n-(2-甲氧基-6-(三氟甲基)苯基)-2-(甲硫基)嘧啶-4-羧酰胺(1.66g,4.39mmol,1当量)中,分数次加入m-cpba(3.03g,17.58mmol,4当量)。混合物室温搅拌16小时后,反应混合物的lcms分析表明完全转化至所需产物。反应混合物经浓缩及硅胶纯化(石油醚:乙酸乙酯=100:0~0:100),获得淡黄色固体的5-氯-n-(2-甲氧基-6-(三氟甲基)苯基)-2-(甲磺酰基)嘧啶-4-羧酰胺(1.19g,收率:66.1%)。
1098、lc-ms m/z:410.01(m+1)+。
1099、步骤3:5-氯-n-(2-甲氧基-6-(三氟甲基)苯基)-2-((4-(4-甲基哌嗪-1-基)-3-(三氟甲基)苯基)氨基)嘧啶-4-羧酰胺
1100、
1101、室温下,在溶于thf(1ml)内的n-(4-(4-甲基哌嗪-1-基)-3-(三氟甲基)苯基)甲酰胺(75.06mg,0.316mmol,1.1当量)内,加入氢化钠(14.64mg,0.366mmol,1.5当量)。随后,混合物在60℃下搅拌0.5小时,获得棕色溶液。搅拌0.5小时后,加入5-氯-n-(2-甲氧基-6-(三氟甲基)苯基)-2-(甲磺酰基)嘧啶-4-羧酰胺(100mg,0.244mmol,1当量)。混合物在氮气气氛中室温搅拌3小时,反应混合物的lcms分析表明完全转化至所需产物。混合物过滤后,滤饼以乙腈洗涤,然后经减压浓缩,去除多余的溶剂。残余物经c18纯化(水:乙腈=90:10~95:05,水/乙腈中溶有0.1%的甲酸)获得实施例184化合物(9.5mg,收率:6.6%)。
1102、lc-ms m/z:589.15(m+1)+;
1103、1h nmr(400mhz,meod):δ8.66(s,1h),8.16(d,j=8.4hz,1h),8.06(d,j=2.4hz,1h),7.57(t,j=7.9hz,1h),7.51(d,j=8.8hz,1h),7.44(d,j=8.2hz,1h),7.38(d,j=7.9hz,1h),3.94(s,3h),3.60(d,j=11.9hz,2h),3.28(d,j=11.5hz,2h),3.21(d,j=7.7hz,4h),3.01(s,3h)。
1104、
1105、
1106、实施例187:
1107、5-氯-n-(2-氯-6-氰基苯基)-2-((3-氟-4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺
1108、
1109、路线图
1110、
1111、步骤1:5-氯-n-(2-氯-6-氰基苯基)-2-(甲硫基)嘧啶-4-羧酰胺
1112、
1113、在溶于dmf(150ml)内的5-氯-2-(甲硫基)嘧啶-4-羧酸(13.41g,65.54mmol,1当量)中,加入diea(25.41g,196.62mmol,3当量)及hatu(32.4g,85.2mmol,1.3当量)。搅拌30分钟后,在反应中加入2-氨基-3-氯苯腈(10g,65.54mmol,1当量)。混合物在21℃下搅拌16小时,获得黑棕色溶液。lcms表明反应未发生。将反应物加热至50℃,并搅拌10小时,获得黑棕色溶液。lcms表明反应起始物质已完全消耗且检测到检测到一个具有所需物质的峰。反应物以水处理,并以乙酸乙酯萃取(60ml×3)。合并有机层经浓氯化铵水溶液及卤水洗涤、硫酸钠干燥、过滤及浓缩,获得棕色固体的5-氯-n-(2-氯-6-氰基苯基)-2-(甲硫基)嘧啶-4-羧酰胺(28g,收率125.96%),并将其直接用于下一步骤。
1114、lc-ms m/z:339.0(m+1)+。
1115、步骤2:5-氯-n-(2-氯-6-氰基苯基)-2-(甲磺酰基)嘧啶-4-羧酰胺
1116、
1117、0~5℃下,在溶于dcm(300ml)内的5-氯-n-(2-氯-6-氰基苯基)-2-(甲硫基)嘧啶-4-羧酰胺(65.54mmol,1当量)中,分批加入85%的m-cpba(39.92g,196.62mmol,3当量,85%纯度)。混合物在22℃下搅拌5小时,获得浅棕色悬浮液。lcms表明起始物质已完全消耗且检测到一个具有所需物质的主峰。反应物倒入冰水中,并以碳酸钠小心碱化至ph=9~10,随后以dcm萃取(150ml×3)。合并有机层经浓碳酸氢钠水溶液(100ml×5)及卤水洗涤、硫酸镁干燥及过滤,并浓缩至干态。残余物经快速柱纯化(二氧化硅,以溶有80%乙酸乙酯的石油醚洗脱,plate 1),获得浅棕色固体的5-氯-n-(2-氯-6-氰基苯基)-2-(甲磺酰基)嘧啶-4-羧酰胺(7.3g,收率30%)。
1118、lc-ms m/z:371.0(m+1)+。
1119、步骤3:n-(3-氟-4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯基)甲酰胺
1120、
1121、将溶于甲酸(30ml)内的3-氟-4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯胺(57.17mmol,1当量)在90℃下搅拌3小时,获得紫色溶液。lcms表明起始物质已完全消耗且检测到具有所需物质的一个新峰。反应物以浓碳酸钠水溶液碱化至ph=10~11,并以dcm/异丙醇=3/1(60ml×3)萃取。合并有机层经卤水洗涤、硫酸镁干燥及过滤后,浓缩至干态。残余物经乙酸乙酯研磨、过滤及油泵内干燥,获得浅棕色固体的n-(3-氟-4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯基)甲酰胺(16.2g,收率88.43%)。
1122、lc-ms m/z:321.2(m+1)+。
1123、步骤4:5-氯-n-(2-氯-6-氰基苯基)-2-((3-氟-4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺
1124、
1125、在溶于dmf(50ml)内的n-(3-氟-4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯基)甲酰胺(3.13g,9.78mmol,1.1当量)中,加入60%的氢化钠(0.71g,17.78mmol,2当量)。40℃下搅拌1小时后,在反应物中加入5-氯-n-(2-氯-6-氰基苯基)-2-(甲磺酰基)嘧啶-4-羧酰胺(3.3g,8.89mmol,1当量)。所得混合物在24℃下搅拌16小时,获得黑棕色溶液。lcms表明起始物质已完全消耗且检测到一个新的峰。在反应物内加入溶于水(20ml)中的氢氧化钠(1.07g,26.67mmol,3当量)后,将反应物继续搅拌1小时,获得黑棕色溶液。lcms表明检测到一个具有所需物质的主峰。反应物以水稀释,并以dcm/异丙醇=3/1(100ml×3)萃取。合并有机层经卤水洗涤、硫酸镁干燥及过滤后,浓缩至干态。残余物经快速柱纯化(二氧化硅,以溶有15%甲醇的dcm洗脱)后,浓缩至干态。所得产物经乙酸乙酯/甲醇研磨及过滤,滤饼在空气中干燥至干态。产物溶于甲醇中,并以溶于甲醇(4ml)内的4m盐酸酸化,获得澄清溶液。浓缩至干态后,残余物经冻干,获得浅棕色固体的实施例187(2332.96mg,收率44.95%)。
1126、lc-ms m/z:583.2(m+1)+。
1127、1h nmr(400mhz,甲醇-d4)δ8.65(s,1h),7.92(dd,j=8.2,1.4hz,1h),7.86(dd,j=7.8,1.4hz,1h),7.80(dd,j=14.7,2.5hz,1h),7.57(t,j=8.0hz,1h),7.48(d,j=8.4hz,1h),7.10(t,j=9.1hz,1h),3.76(d,j=12.2hz,4h),3.71~3.65(m,1h),3.61(d,j=13.9hz,2h),3.41(t,j=12.0hz,2h),3.30~3.14(m,4h),2.95(s,3h),2.57(d,j=13.5hz,2h),2.24~2.11(m,2h)。
1128、
1129、实施例189:
1130、5-氯-n-(2-氰基-6-甲氧基苯基)-2-((6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-羧酰胺
1131、
1132、路线图
1133、
1134、步骤1:n-(6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)甲酰胺
1135、
1136、将6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-胺(3.5g,18.20mmol,1.00当量)与甲酸(30ml)的混合物回流搅拌4小时,获得淡黄色溶液。反应混合物的lc-ms分析表明完全转化至所需产物。反应混合物在真空中浓缩至干态后,残余物经快速柱纯化(二氧化硅,以溶有40%~60%甲醇的乙酸乙酯洗脱,combiflash nextgen 300),获得米白色固体的n-(6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)甲酰胺化合物(1.50g,收率:38%)。
1137、lc-ms m/z:221.2(m+1)+。
1138、步骤2:5-氯-2-(n-(6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)甲酰胺基)嘧啶-4-羧酸甲酯
1139、
1140、在溶于thf(30ml)内的n-(6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)甲酰胺(0.89g,4.03mmol,1.0当量)中,分三批加入氢化钠(0.24g,6.05mmol,1.50当量)。50℃下搅拌0.5小时后,获得灰色悬浮液,随后加入5-氯-2-(甲磺酰基)嘧啶-4-羧酸甲酯(1.79g,6.05mmol,1.5当量)。混合物在氮气气氛中50℃搅拌4小时,获得棕色悬浮液。反应混合物的lcms分析表明大部分转化至所需产物。混合物内加入乙酸(1ml)后,进行过滤。滤饼经thf(50ml)洗涤,随后经减压浓缩,去除多余的溶剂。残余物经快速柱纯化(二氧化硅,以溶有45%~55%甲醇的乙酸乙酯洗脱,combiflash nextgen 300),获得黄色固体的5-氯-2-(n-(6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)甲酰胺基)嘧啶-4-羧酸酯化合物(0.62g,收率:39%)。
1141、lc-ms m/z:391.8(m+1)+。
1142、步骤3:5-氯-2-((6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-羧酸
1143、
1144、在溶于1,4-二恶烷(6ml)内的5-氯-2-(n-(6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)甲酰胺基)嘧啶-4-羧酸酯(0.62g,1.59mmol,1.00当量)中,加入溶于1,4-二恶烷(6ml)中的4n盐酸,并室温搅拌2小时,获得黄色悬浮液。反应混合物的lcms分析表明完全转化至所需产物。随后,反应混合物经减压浓缩,去除多余的溶剂,获得黄色固体的中间产物。
1145、在溶于甲醇(5ml)和水(5ml)内的中间产物中,加入氢氧化钠(0.4g),并搅拌2小时,获得棕色溶液。反应混合物的lcms分析表明完全转化至所需产物。加入柠檬酸(1.3g)后,经减压浓缩,去除多余的溶剂。所得残余物经快速柱纯化(c18,0~100%a(水+0.3%tfa),40%~50%b(accn+0.3%tfa),combiflash nextgen300),获得黄色固体的5-氯-2-((6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-羧酸化合物(250mg,收率:45%)。
1146、lc-ms m/z:349.8(m+1)+。
1147、步骤4:5-氯-n-(2-氰基-6-甲氧基苯基)-2-((6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-羧酰胺
1148、
1149、在混于吡啶(5ml)内的5-氯-2-((6-甲氧基-2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-羧酸(100mg,0.29mmol,1.0当量)及2-氨基-3-甲氧基苯腈(86mg,0.58mmol,4.00当量)中,加入三氯氧磷(178mg,1.16mmol,4.00当量)。混合物在25℃下搅拌1小时,获得黑色溶液。反应混合物的lcms分析表明反应完成。混合物倒入饱和碳酸氢钠溶液(50ml)中后,以乙酸乙酯(20ml×2)萃取。有机层经硫酸镁干燥后,在真空中浓缩至干态。残余物经快速柱纯化(c18,0~100%a(水+0.3%tfa),60%~65%b(accn+0.3%tfa),combiflashnextgen 300),获得棕色固体的实施例189化合物(67mg,收率:45%,三氟乙酸盐)。
1150、lc-ms m/z:479.94(m+1)+;
1151、1h nmr(400mhz,meod)δ8.62(d,j=8.0hz,1h),8.31(s,1h),7.62~7.31(m,3h),6.94(s,1h),4.59(d,j=14.8hz,1h),4.29(d,j=14.8hz,1h),3.97(s,3h),3.96(s,3h),3.75(s,1h),3.41(s,1h),3.25(d,j=8.1hz,1h),3.14(d,j=14.4hz,1h),3.06(s,3h)。
1152、
1153、
1154、实施例192:
1155、n-(2-氯-6-甲氧基苯基)-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)-5-乙烯基嘧啶-4-羧酰胺
1156、
1157、路线图
1158、
1159、步骤1:5-溴-2-(甲硫基)嘧啶-4-羧酸甲酯
1160、
1161、0℃下,在溶于甲醇(300ml)内的5-溴-2-(甲硫基)嘧啶-4-羧酸(30.0g,120.44mmol,1当量)中,缓慢逐滴加入氯化亚砜(22ml)。随后,混合物在60℃下搅拌1小时,获得棕色溶液。lcms分析表明检测到所需物质且反应完成。混合物经减压浓缩,去除多余的溶剂。残余物在搅拌的同时倒入碳酸氢钠水溶液(200ml)中,并以dcm(200ml×2)萃取。有机相经卤水洗涤、硫酸钠干燥及浓缩,获得橙色固体的5-溴-2-(甲硫基)嘧啶-4-羧酸甲酯(29.3g,92.5%)。
1162、lc-ms m/z:264.11(m+1)+。
1163、步骤2:5-溴-2-(甲磺酰基)嘧啶-4-羧酸甲酯
1164、
1165、在溶于二氯甲烷(1000ml)内的5-溴-2-(甲硫基)嘧啶-4-羧酸甲酯(29.3g,111.36mmol,1当量)中,分数次加入m-cpba(79.13g,389.76mmol,3.5当量)。混合物室温搅拌16小时后,反应混合物的lcms分析表明完全转化至所需产物。反应混合物过滤后,滤过物以饱和碳酸氢钠溶液(500ml)洗涤。有机层经硫酸镁干燥后,在真空中浓缩至干态。残余物经硅胶纯化(石油醚:乙酸乙酯=80:20~60:40),获得淡黄色固体的5-溴-2-(甲磺酰基)嘧啶-4-羧酸甲酯(36.5g,收率:111.1%)。
1166、lc-ms m/z:296.11(m+1)+。
1167、步骤3:5-溴-2-(n-(4-(4-甲基哌嗪-1-基)苯基)甲酰胺基)嘧啶-4-羧酸甲酯
1168、
1169、室温下,在溶于thf(70ml)内的n-(4-(4-甲基哌嗪-1-基)苯基)甲酰胺(6g,27.36mmol,1当量)中,分批加入氢化钠(1.64g,41.04mmol,1.5当量)。随后,混合物在60℃下搅拌1小时,获得棕色溶液。搅拌0.5小时后,加入5-溴-2-(甲磺酰基)嘧啶-4-羧酸甲酯(16.15g,54.72mmol,2当量)。混合物在氮气气氛中室温搅拌14小时。反应混合物的lcms分析表明完全转化至所需产物。混合物过滤后,滤饼以thf(200ml)洗涤,然后经减压浓缩,去除多余的溶剂。残余物经硅胶纯化(二氯甲烷:甲醇=100:0~90:10),获得5-溴-2-(n-(4-(4-甲基哌嗪-1-基)苯基)甲酰胺基)嘧啶-4-羧酸甲酯(2.25g,收率:18.9%)。
1170、lc-ms m/z:434.07(m+1)+。
1171、步骤4:5-溴-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-羧酸
1172、
1173、在混于1,4-二恶烷(20ml)内的5-溴-2-(n-(4-(4-甲基哌嗪-1-基)苯基)甲酰胺基)嘧啶-4-羧酸甲酯中,加入溶于1,4-二恶烷(3ml)内的4n盐酸。混合物室温搅拌48小时后,进一步加入5ml溶于1,4-二恶烷内的4n盐酸。反应混合物的lcms分析表明完全转化至所需产物。随后,反应物经减压浓缩,去除多余的溶剂。残余物中加入10%氢氧化钠水溶液,并搅拌0.5小时。反应混合物的lcms分析表明完全转化至所需产物。残余物中加入柠檬酸(3.4g)和水(20ml),并进行过滤。滤饼以thf洗涤,并经真空干燥,获得棕色固体的5-溴-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-羧酸(1.66g,收率:18.9%)。
1174、lc-ms m/z:392.06(m+1)+。
1175、步骤5:5-溴-n-(2-氯-6-甲氧基苯基)-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺
1176、
1177、在混于吡啶内的5-溴-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-羧酸(1.57g,4.0mmol,1.0当量)及2-氯-6-甲氧基苯胺(1.65g,6.0mmol,1.5当量)中,加入三氯氧磷(1.5ml,16.01mmol,4当量)。混合物以氮气脱气3次,并在氮气气氛中室温搅拌4小时。反应混合物的lcms分析表明完全转化至所需产物。反应混合物倒入碳酸氢钠水溶液(200ml),并经乙酸乙酯(100ml×3)过滤及减压浓缩,获得粗产物。粗产物经硅胶纯化(石油醚:乙酸乙酯=100:0~60:40),获得黄色固体的5-溴-n-(2-氯-6-甲氧基苯基)-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺(1.43g,67.2%)。
1178、lc-ms m/z:531.08(m+1)+。
1179、步骤6:n-(2-氯-6-甲氧基苯基)-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)-5-乙烯基嘧啶-4-羧酰胺
1180、
1181、将溶于1,4-二恶烷(1ml)中的5-溴-n-(2-氯-6-甲氧基苯基)-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺(100mg,0.188mmol,1当量)、4,4,5,5-四甲基-2-乙烯基-1,3,2-二氧硼杂环戊烷(34.75mg,0.226mmol,1.2当量)、磷酸钾(99.78mg,0.470mmol,2.5当量)、[1,1'-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物(pd(dppf)cl2 dcm)(15.35mg,0.019mmol,0.1当量)通过微波反应器加热至125℃,并搅拌1小时。反应混合物的lcms分析表明完全转化至所需产物。混合物经减压浓缩,去除多余的溶剂。残余物经硅胶纯化(二氯甲烷:甲醇=100:0~90:10),获得粗产物。粗产物经制备型tlc进一步纯化,获得黄色固体的实施例192(18.4mg,20%)。
1182、lc-ms m/z:479.19(m+1)+;
1183、1h nmr(400mhz,meod):δ8.82(s,1h),7.64(d,j=9.0hz,2h),7.42(dd,j=17.8,11.2hz,1h),7.34(t,j=8.3hz,1h),7.17~7.12(m,1h),7.09(d,j=8.4hz,1h),7.01(d,j=9.0hz,3h),5.74(d,j=17.7hz,1h),5.28(d,j=12.0hz,1h),3.91(s,3h),3.25(d,j=4.2hz,6h),2.84(s,5h),2.51(s,4h)。
1184、实施例193:
1185、7-(2-氯-6-甲氧基苯基)-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)吡啶并[3,4-d]嘧啶-8(7h)-酮
1186、
1187、路线图
1188、
1189、110℃下,将5-溴-n-(2-氯-6-甲氧基苯基)-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺(250mg,0.47mmol,1.00当量)、(e)-2-(2-乙氧基乙烯基)-4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷(186mg,0.94mmol,2.00当量)、pd(dppf)cl2 dcm(41mg,0.05mmol,0.1当量)、磷酸钾(195mg,0.94mmol,2.00当量)、1,4-二恶烷(3ml)及水(0.5ml)的混合物微波搅拌2小时。反应混合物的lc-ms分析表明完全转化至所需产物。混合物倒入水中(20ml),并以乙酸乙酯(50ml)萃取。有机层经硫酸镁干燥后,在真空中浓缩至干态。
1190、所得残余物中加入thf(20ml)及盐酸(30%)(2.5ml),且室温搅拌2小时。反应混合物的lc-ms分析表明完全转化至所需产物。反应混合物在真空中浓缩至干态,并经快速柱纯化(c18,0~100%a(水+0.01%tfa),40%~50%b(乙腈+0.01%tfa),combiflash nextgen300)获得棕色固体的实施例193化合物(65mg,收率:29%(三氟乙酸盐))。
1191、lc-ms m/z:478.0(m+1)+。
1192、1h nmr(400mhz,meod)δ7.54(t,j=8.4hz,1h),7.37(t,j=6.1hz,2h),7.32(dd,j=7.1,0.9hz,1h),7.27~7.18(m,4h),6.56(d,j=6.8hz,1h),5.74(d,j=5.1hz,1h),3.98(d,j=13.8hz,2h),3.87(d,j=13.0,4.4hz,3h),3.67(d,j=12.5hz,2h),3.29(s,2h),3.17(t,j=12.0hz,2h),3.00(d,j=2.6hz,3h)。
1193、实施例194:
1194、n-(2-氟-6-羟基苯基)-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)-5-乙烯基嘧啶-4-羧酰胺
1195、
1196、路线图
1197、
1198、步骤1:n-(2-(烯丙氧基)-6-氟苯基)-5-溴-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺
1199、
1200、在溶于dmf(3ml)内的5-溴-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-羧酸(222mg,1.33mmol,1.00当量)中,加入sm2(520mg,1.33mmol,1.00当量)及2-(烯丙氧基)-6-氟苯胺hatu(506mg,1.33mmol,1.00当量),60℃下搅拌3小时,获得棕色溶液。lc-ms表明获得目标化合物ms。混合物倒入水中(20ml),并以乙酸乙酯萃取(20ml×2)。有机层经硫酸钠干燥后,在真空中浓缩至干态,获得710mg的棕色固体,并将其直接用于下一反应。
1201、lc-ms m/z:542.4(m+1)+。
1202、步骤2:n-(2-氟-6-羟基苯基)-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)-5-乙烯基嘧啶-4-羧酰胺
1203、
1204、将n-(2-(烯丙氧基)-6-氟苯基)-5-溴-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺(720mg,1.33mmol,1.00当量)、4,4,5,5-四甲基-2-乙烯基-1,3,2-二氧硼杂环戊烷(410mg,2.66mmol,2.00当量)、pd(dppf)cl2 dcm(106mg,0.13mmol,0.1当量)、磷酸钾(565mg,2.66mmol,2.00当量)、1,4-二恶烷(10ml)及水(1ml)的混合物在100℃下微波搅拌1小时,获得黑色悬浮液。反应混合物的lc-ms分析表明完全转化至所需产物。混合物倒入水中(25ml),并以乙酸乙酯(30ml)萃取。有机层经硫酸钠干燥后,在真空中浓缩至干态。残余物经快速柱纯化(c18,0~100%a(水+0.3%tfa),55%~65%b(accn+0.3%tfa),combiflash nextgen 300),获得黄色固体的实施例194化合物(230mg,收率:38%,三氟乙酸盐)。
1205、lc-ms m/z:449.5(m+1)+;
1206、1h nmr(400mhz,meod)δ8.83(d,j=10.8hz,1h),7.80~7.59(m,2h),7.57~7.39(m,1h),7.18(td,j=8.3,6.4hz,1h),7.09~7.00(m,2h),6.83~6.69(m,2h),5.71(t,j=18.2hz,1h),5.28(t,j=15.0hz,1h),3.80(d,j=12.3hz,2h),3.61(s,2h),3.30~3.21(m,2h),3.06(d,j=11.3hz,2h),3.02(s,j=18.2hz,3h)。
1207、实施例195:
1208、5-氯-n-(2-氰基-6-氟苯基)-2-((3-氟-4-(4-(1-羟基-2-甲基丙烷-2-基)哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺
1209、
1210、路线图
1211、
1212、步骤1:5-氯-n-(2-氰基-6-氟苯基)-2-((3-氟-4-(4-(2-甲基-1-氧代丙烷-2-基)哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺
1213、
1214、将5-氯-n-(2-氰基-6-氟苯基)-2-((3-氟-4-(哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺(50mg,0.11mmol,1.00当量)、2-溴-2-甲基丙烷(32mg,0.22mmol,1.50当量)、三乙胺(10ml)及碳酸钾(30mg,0.22mmol,2.0当量)的混合物在70℃下搅拌3小时。lc-ms检测到目标化合物。混合物过滤后,滤过物在真空中浓缩至干态,获得55mg的黄色固体,直接用于下一反应。
1215、步骤2:5-氯-n-(2-氰基-6-氟苯基)-2-((3-氟-4-(4-(1-羟基-2-甲基丙烷-2-基)哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺
1216、
1217、将5-氯-n-(2-氰基-6-氟苯基)-2-((3-氟-4-(4-(2-甲基-1-氧代丙烷-2-基)哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺(55mg,0.10mmol,1.00当量)、硼氢化钠(11mg,0.30mmol,3.00当量)及乙醇(10ml)的混合物在25℃下搅拌3小时。lc-ms检测到目标化合物。混合物经减压浓缩,去除多余的溶剂。残余物中加入水(5ml)后,以乙酸乙酯(10ml×2)萃取。合并有机层经硫酸钠干燥后,在真空中浓缩至干态,随后经快速柱纯化(c18,0~100%a(水+1%甲酸),40%~50%b(accn+1%甲酸),combiflash nextgen 300),获得黄色固体的实施例195化合物(20mg,收率:37%,甲酸盐)。
1218、lc-ms m/z:541.0(m+1)+;
1219、1h nmr(400mhz,meod)δ8.65(s,1h),7.75~7.56(m,4h),7.52(d,j=8.0hz,1h),7.10(t,j=9.2hz,1h),3.70(d,j=16.0hz,3h),3.53(m,6h),3.20(t,j=11.6hz,2h),1.44
1220、(s,6h)。
1221、实施例196:
1222、5-氯-n-(2-氰基-6-甲氧基苯基)-2-((4-(4-(2-甲基-1-氧代丙烷-2-基)哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺
1223、
1224、路线图
1225、
1226、步骤1:5-氯-n-(2-氰基-6-甲氧基苯基)-2-((4-(4-(1-羟基-2-甲基丙烷-2-基)哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺
1227、
1228、将5-氯-n-(2-氰基-6-甲氧基苯基)-2-((4-(哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺(80mg,0.17mmol,1.00当量)、2-溴-2-甲基丙烷(51mg,0.34mmol,2.00当量)、乙腈(10ml)及碳酸钾(47mg,0.34mmol,2.0当量)的混合物在70℃下搅拌3小时。lc-ms表明检测到目标化合物ms。混合物过滤后,滤过物在真空中浓缩至干态后,获得85mg的黄色固体,直接用于下一反应。
1229、lc-ms m/z:535.0(m+1)+。
1230、步骤2:5-氯-n-(2-氰基-6-甲氧基苯基)-2-((4-(4-(2-甲基-1-氧代丙烷-2-基)哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺
1231、
1232、将5-氯-n-(2-氰基-6-甲氧基苯基)-2-((4-(4-(1-羟基-2-甲基丙烷-2-基)哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺(85mg,0.16mmol,1.00当量)、硼氢化钠(18mg,0.48mmol,3.00当量)及乙醇(10ml)的混合物在25℃下搅拌3小时,获得淡黄色溶液。lc-ms表明检测到目标化合物ms。混合物经减压浓缩,去除多余的溶剂。残余物中加入水(5ml)后,以乙酸乙酯(10ml×2)萃取。合并有机层经硫酸钠干燥后,在真空中浓缩至干态,并经快速柱纯化(c18,0~100%a(水+1%甲酸),40%~50%b(accn+1%甲酸),combiflash nextgen 300),获得棕色固体的实施例196化合物(23mg,收率:27%,甲酸盐)。
1233、lc-ms m/z:537.0;
1234、1h nmr(400mhz,meod)δ8.56(s,j=3.5hz,1h),7.65(d,j=8.6hz,2h),7.54~7.44(m,2h),7.41(d,j=7.3hz,1h),7.03(d,j=8.9hz,2h),4.00(d,j=34.0hz,3h),3.61(s,2h),3.15(s,8h),1.29(s,6h)。
1235、实施例197:
1236、2-(4-(4-((5-氯-4-((2,6-二氯苯基)氨基甲酰基)嘧啶-2-基)氨基)苯基)哌嗪-1-基)乙酸
1237、
1238、路线图
1239、
1240、步骤1:1-甲基-4-(1-(2-甲基-4-亚硝基苯基)哌啶-4-基)哌嗪
1241、
1242、将溶于甲酸(30ml)中的3-甲基-4-(4-(4-甲基哌嗪-1-基)哌啶-1-基)苯胺(4.51g,15.64mmol,1当量)加热至80℃,并搅拌1.5小时,获得棕色溶液。tlc表明检测到新的斑点,而且反应混合物的lcms分析表明完全转化至所需产物。冷却至室温后,反应混合物经浓缩、dcm稀释及卤水洗涤。分离后的有机相经干燥硫酸钠干燥和浓缩,获得白色固体的1-甲基-4-(1-(2-甲基-4-亚硝基苯基)哌啶-4-基)哌嗪(3.39g,收率:68.5%)。
1243、lc-ms m/z:303.21(m+1)+。
1244、步骤2:5-氯-2-(n-(3-甲基-4-(4-(4-甲基哌嗪-1-基)哌啶-1-基)苯基)甲酰胺基)嘧啶-4-羧酸甲酯
1245、
1246、室温下,在溶于dmf(30ml)内的1-甲基-4-(1-(2-甲基-4-亚硝基苯基)哌啶-4-基)哌嗪(3.19g,10.08mmol,1当量)中,加入氢化钠(550mg,13.75mmol,1.5当量)。随后,将混合物在室温下搅拌0.5小时,获得紫色溶液。搅拌0.5小时后,加入5-氯-2-(甲磺酰基)嘧啶-4-羧酸甲酯(2.3g,9.16mmol,1当量)。混合物在氮气气氛中室温搅拌3小时,获得橙色溶液。反应混合物的lcms分析表明完全转化至所需产物。混合物过滤后,滤饼以dcm洗涤,随后经减压浓缩,去除多余的溶剂。残余物经c18纯化(水:乙腈=90:10~95:05,水/乙腈中溶有0.1%的甲酸),获得橙色液体的5-氯-2-(n-(3-甲基-4-(4-(4-甲基哌嗪-1-基)哌啶-1-基)苯基)甲酰胺基)嘧啶-4-羧酸甲酯(3.71g,收率:88.2%)。
1247、lc-ms m/z:487.21(m+1)+。
1248、步骤3:5-氯-2-((3-甲基-4-(4-(4-甲基哌嗪-1-基)哌啶-1-基)苯基)氨基)嘧啶-4-羧酸
1249、
1250、在溶于乙醇/水=5:1(30ml)内的5-氯-2-(n-(3-甲基-4-(4-(4-甲基哌嗪-1-基)哌啶-1-基)苯基)甲酰胺基)嘧啶-4-羧酸甲酯(3.71g,7.62mmol,1当量)中,加入氢氧化钠水溶液(5ml,22.85mmol,3当量)。随后,混合物在室温下搅拌16小时,获得黄色溶液。反应混合物的lcms分析表明完全转化至所需产物。反应混合物经浓缩,去除乙醇,并随后加入柠檬酸(1.2g)和水(5ml)。混合物过滤后,滤饼以水洗涤,并经干燥,获得白色固体的5-氯-2-((3-甲基-4-(4-(4-甲基哌嗪-1-基)哌啶-1-基)苯基)氨基)嘧啶-4-羧酸(2.23g,收率:61.6%)。
1251、lc-ms m/z:445.20(m+1)+。
1252、步骤4:2-(4-(4-((5-氯-4-((2,6-二氯苯基)氨基甲酰基)嘧啶-2-基)氨基)苯基)哌嗪-1-基)乙酸
1253、
1254、在混于乙腈(1ml)内的5-氯-2-((3-甲基-4-(4-(4-甲基哌嗪-1-基)哌啶-1-基)苯基)氨基)嘧啶-4-羧酸(100mg,0.225mmol,1当量)及2-氨基-3-氟苯腈(30.59mg,0.225mmol,1当量)中,加入三氯氧磷(0.084ml,0.899mmol,4当量)。混合物在氮气气氛中室温搅拌1小时,获得棕色溶液。反应混合物的lcms分析表明完全转化至所需产物。反应物中加入碳酸氢钠水溶液,以调节至ph=7。过滤后,残余物经c18纯化(水:乙腈=90:10~95:05,水/乙腈中溶有0.1%的甲酸),获得橙色固体的实施例197化合物(33.1mg,收率:26.2%)。
1255、lc-ms m/z:563.24(m+1)+。
1256、1h nmr(400mhz,meod)δ8.58(s,1h),7.71(d,j=7.5hz,1h),7.68~7.53(m,3h),7.49(d,j=2.4hz,1h),7.06(d,j=8.7hz,1h),3.23~2.66(m,13h),2.62(s,3h),2.32(s,3h),2.05(d,j=11.5hz,2h),1.77(dt,j=11.7,8.1hz,2h)。
1257、
1258、
1259、实施例200:
1260、5-氯-n-(2-氯-6-氟苯基)-2-((3-氟-4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺
1261、
1262、路线图
1263、
1264、步骤1:1-(2-氟-4-亚硝基苯基)-4-甲基哌嗪
1265、
1266、以甲酸(15ml)处理3-氟-4-(4-甲基哌嗪-1-基)苯胺(2.09g,9.99mmol,1当量)后,将反应物在110℃下加热5小时。反应混合物的lcms分析表明完全转化至所需产物。冷却至室温后,反应混合物经减压浓缩,并通过以碳酸氢钠处理而调至ph=7~8。残余物经干燥及有机相洗涤后,有机相经浓缩,获得浅紫色固体的1-(2-氟-4-亚硝基苯基)-4-甲基哌嗪(2.74g,收率:115.8%)。
1267、lc-ms m/z:210.13(m+1)+。
1268、步骤2:5-氯-n-(2-氯-6-氟苯基)-2-(n-(3-氟-4-(4-甲基哌嗪-1-基)苯基)甲酰胺基)嘧啶-4-羧酰胺
1269、
1270、室温下,在溶于thf(1ml)内的1-(2-氟-4-亚硝基苯基)-4-甲基哌嗪(71.67mg,0.302mmol,1.1当量)中,加入氢化钠(16.47mg,0.412mmol,1.5当量)。随后,混合物在室温下搅拌0.5小时,获得棕色溶液。搅拌0.5小时后,加入5-氯-n-(2-氯-6-氟苯基)-2-(甲磺酰基)嘧啶-4-羧酰胺(100mg,0.275mmol,1当量)。混合物在氮气气氛中室温搅拌3小时。反应混合物的lcms分析表明完全转化至所需产物。混合物过滤后,滤饼经dcm洗涤(50ml),然后经减压浓缩,去除多余的溶剂。残余物经硅胶纯化(二氯甲烷:甲醇=100:0~95:5),获得黄色固体的5-氯-n-(2-氯-6-氟苯基)-2-(n-(3-氟-4-(4-甲基哌嗪-1-基)苯基)甲酰胺基)嘧啶-4-羧酰胺(34.4mg,收率:24.4%)。
1271、lc-ms m/z:521.10(m+1)+。
1272、步骤3:5-氯-n-(2-氯-6-氟苯基)-2-((3-氟-4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺
1273、
1274、在5-氯-n-(2-氯-6-氟苯基)-2-(n-(3-氟-4-(4-甲基哌嗪-1-基)苯基)甲酰胺基)嘧啶-4-羧酰胺(34.4mg,0.065mmol,1当量)的混合物中,加入4n盐酸/1,4-二恶烷(1ml)。混合物在室温下搅拌2小时后,反应混合物的lcms分析表明完全转化至所需产物。随后,混合物经过滤和浓缩,获得粗产物。粗产物经c18纯化(水:乙腈=90:10~95:05,水/乙腈中溶有0.1%的甲酸),获得黄色固体的实施例200化合物(16.5mg,收率:51.3%)。
1275、lc-ms m/z:493.10(m+1)+
1276、1h nmr(400mhz,meod):δ8.63(s,1h),7.83(dd,j=14.8,2.4hz,1h),7.44~7.37(m,3h),7.31~7.23(m,1h),7.08(t,j=9.2hz,1h),3.59(dd,j=21.8,12.7hz,4h),3.70~3.47(m,4h),3.12(t,j=12.4hz,2h),3.00(s,3h)。
1277、
1278、
1279、实施例203:
1280、(s)-5-氯-n-(2-氰基-6-甲氧基苯基)-2-((3-甲基-1,2,3,4,4a,5-六氢苯并[b]吡嗪并[1,2-d][1,4]恶嗪-8-基)氨基)嘧啶-4-羧酰胺
1281、
1282、路线图
1283、
1284、步骤1:(s)-8-硝基-1,2,4a,5-四氢苯并[b]吡嗪并[1,2-d][1,4]恶嗪-3(4h)-羧酸叔丁酯
1285、
1286、在混于dmso(20ml)内的(s)-3-(羟基甲基)哌嗪-1-羧酸叔丁酯(2g,8.61mmol,1当量)中,加入1,2-二氟-4-硝基苯(1.78g,11.19mmol,1.3当量)及氢氧化钾(1.45g,25.83mmol,3当量)。反应物在18℃下搅拌3小时,获得棕色溶液。lcms表明(s)-3-(羟基甲基)哌嗪-1-羧酸叔丁酯完全消耗。反应物加热至60℃,并继续搅拌8小时,获得棕色溶液。lcms表明大约一半的起始物质转化至所需产物。反应物加热至70℃,并继续搅拌8小时,获得棕色溶液。lcms表明大部分起始物质已被消耗。反应物加热至80℃,并继续搅拌3小时,获得棕色溶液。lcms表明反应完成。反应物以水稀释,沉淀出大量固体。过滤且以水洗涤后,滤饼在油泵中干燥,获得黄色固体的(s)-8-硝基-1,2,4a,5-四氢苯并[b]吡嗪并[1,2-d][1,4]恶嗪-3(4h)-羧酸叔丁酯(2.88g,收率95.05%)。
1287、lc-ms m/z:336.2(m+1)+。
1288、步骤2:(s)-3-甲基-8-硝基-1,2,3,4,4a,5-六氢苯并[b]吡嗪并[1,2-d][1,4]恶嗪
1289、
1290、氮气气氛中,在溶于甲酸(3.07ml,81.96mmol,10当量)内的(s)-8-硝基-1,2,4a,5-四氢苯并[b]吡嗪并[1,2-d][1,4]恶嗪-3(4h)-羧酸叔丁酯(5g,8.2mmol,1当量)中,加入38%的甲醛水溶液(6ml,81.96mmol,10当量)。反应物在100℃下搅拌4小时,获得棕色溶液。lcms表明起始物质已完全消耗且检测到一个具有所需物质的主峰。反应物以浓碳酸钠水溶液碱化,并以乙酸乙酯萃取(30ml×3)。合并有机层经卤水洗涤、硫酸钠干燥、过滤及浓缩,获得黄色固体的(s)-3-甲基-8-硝基-1,2,3,4,4a,5-六氢苯并[b]吡嗪并[1,2-d][1,4]恶嗪(2.15g,收率105.39%)。
1291、lc-ms m/z:250.1(m+1)+。
1292、步骤3:(s)-3-甲基-1,2,3,4,4a,5-六氢苯并[b]吡嗪并[1,2-d][1,4]恶嗪-8-胺
1293、
1294、氮气气氛中,在溶于甲醇(20ml)内的(s)-3-甲基-8-硝基-1,2,3,4,4a,5-六氢苯并[b]吡嗪并[1,2-d][1,4]恶嗪(8.2mmol,1当量)中,加入钯碳(0.15g)。反应物以氢气脱气数次,并在19℃下搅拌16小时,获得黑棕色悬浮液。lcms表明起始物质已完全消耗且检测到一个具有所需物质的主峰。反应物以硅藻土垫过滤后,滤饼以甲醇洗涤。滤过物经浓缩后,获得浅棕色固体的(s)-3-甲基-1,2,3,4,4a,5-六氢苯并[b]吡嗪并[1,2-d][1,4]恶嗪-8-胺(1.9g,收率105.56%)。
1295、lc-ms m/z:220.2(m+1)+。
1296、步骤4:(s)-n-(3-甲基-1,2,3,4,4a,5-六氢苯并[b]吡嗪并[1,2-d][1,4]恶嗪-8-基)甲酰胺
1297、
1298、将溶于甲酸(10ml)内的(s)-3-甲基-1,2,3,4,4a,5-六氢苯并[b]吡嗪并[1,2-d][1,4]恶嗪-8-胺(1.5g,6.84mmol,1当量)在80℃下搅拌3小时,获得黑棕色溶液。lcms表明起始物质已完全消耗和检测到具有所需物质的一个新峰。反应物以浓碳酸氢钠水溶液碱化至ph=10~11,并以dcm/异丙醇=3/1(20ml×3)萃取。合并有机层经卤水洗涤、硫酸镁干燥、过滤及浓缩,获得灰色固体的(s)-n-(3-甲基-1,2,3,4,4a,5-六氢苯并[b]吡嗪并[1,2-d][1,4]恶嗪-8-基)甲酰胺(1.62g,收率95.86%)。
1299、lc-ms m/z:248.2(m+1)+。
1300、步骤5:(s)-5-氯-2-((3-甲基-1,2,3,4,4a,5-六氢苯并[b]吡嗪并[1,2-d][1,4]恶嗪-8-基)氨基)嘧啶-4-羧酸
1301、
1302、在溶于dmf(5ml)内的(s)-n-(3-甲基-1,2,3,4,4a,5-六氢苯并[b]吡嗪并[1,2-d][1,4]恶嗪-8-基)甲酰胺(400mg,1.62mmol,1当量)中,加入60%的氢化钠(129.39mg,3.23mmol,2当量)。40℃下搅拌0.5小时后,反应物冷却至0~5℃,并加入5-氯-2-(甲磺酰基)嘧啶-4-羧酸甲酯(608.14mg,2.43mmol,1.5当量)。所得混合物搅拌16小时,获得棕色悬浮液。lcms表明起始物质已完全消耗且检测到一个新的峰。反应物中加入溶于水(5ml)中的氢氧化钠(194.08mg,4.85mmol,3当量),并继续搅拌2小时,获得棕色溶液。lcms表明检测到一个具有所需物质的主峰。反应物以水稀释,并以dcm洗涤(10ml)。水层以1n盐酸水溶液酸化至ph=2~3,并以dcm/异丙醇=3/1(10ml×3)萃取。合并有机层经卤水洗涤、硫酸镁干燥及过滤后,浓缩至干态。残余物以乙腈研磨,并进行过滤。滤饼冻干后,获得浅棕色固体的(s)-5-氯-2-((3-甲基-1,2,3,4,4a,5-六氢苯并[b]吡嗪并[1,2-d][1,4]恶嗪-8-基)氨基)嘧啶-4-羧酸(451mg,收率74.19%)。
1303、lc-ms m/z:376.2(m+1)+。
1304、步骤5:(s)-5-氯-n-(2-氰基-6-甲氧基苯基)-2-((3-甲基-1,2,3,4,4a,5-六氢苯并[b]吡嗪并[1,2-d][1,4]恶嗪-8-基)氨基)嘧啶-4-羧酰胺
1305、
1306、0℃下,在溶于吡啶(2ml)内的2-氨基-3-甲氧基苯腈(50mg,337.46μmol,1当量)及(s)-5-氯-2-((3-甲基-1,2,3,4,4a,5-六氢苯并[b]吡嗪并[1,2-d][1,4]恶嗪-8-基)氨基)嘧啶-4-羧酸(126.82mg,337.46μmol,1当量)中,加入三氯氧磷(155.22mg,1.01mmol,3当量)。反应物在21℃下搅拌0.5小时,获得棕色溶液。lcms表明起始物质已完全消耗且检测到一个具有所需物质的峰。反应物以浓碳酸氢钠水溶液碱化至ph=9~10,并以dcm/异丙醇=3/1(10ml×3)萃取。合并有机层经卤水洗涤、硫酸镁干燥及过滤后,浓缩至干态。残余物经快速柱纯化(c18,以含0.1%甲酸的52%乙腈水溶液洗脱),而且流出物经冻干,获得淡黄色固体的实施例203(21.22mg,收率12.43%)。
1307、lc-ms m/z:506.2(m+1)+;
1308、1h nmr(400mhz,甲醇-d4)δ8.55(s,1h),7.54~7.43(m,2h),7.40(dd,j=7.3,1.7hz,1h),7.22(d,j=2.5hz,1h),7.22~7.14(m,1h),6.87(d,j=8.9hz,1h),4.26(dd,j=10.7,2.6hz,1h),4.05~3.95(m,1h),3.96(s,3h),3.87~3.80(m,1h),3.23~3.09(m,2h),3.04(d,j=11.3hz,1h),2.86~2.75
1309、(m,1h),2.52(d,j=8.2hz,1h),2.50(s,3h),2.13(t,j=11.1hz,1h)。
1310、
1311、
1312、实施例206:
1313、5-氯-n-(2-氰基-6-甲氧基苯基)-2-((3-甲氧基-4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺
1314、
1315、路线图
1316、
1317、步骤1:5-氯-2-(甲硫基)嘧啶-4-羧酸甲酯
1318、
1319、在溶于甲醇(350ml)内的5-氯-2-(甲硫基)嘧啶-4-羧酸(50g,244.35mmol,1当量)中,逐滴加入二氯亚砜(43.6g,366.52mmol,1.5当量)。混合物在60℃下搅拌2小时,获得棕色悬浮液。lcms表明起始物质已完全消耗且检测到一个具有所需物质的峰。两项反应并行进行,随后合于一起,以供后处理。反应以冰水淬灭,并通过浓缩去除大部分溶剂。混合物以水稀释,并以乙酸乙酯萃取(500ml×3)。合并有机层经卤水洗涤、硫酸钠干燥、过滤及浓缩,获得棕色油状物的5-氯-2-(甲硫基)嘧啶-4-羧酸甲酯(94.8g,收率88.71%)。
1320、lc-ms m/z:219.1(m+1)+。
1321、步骤2:5-氯-2-(甲磺酰基)嘧啶-4-羧酸甲酯
1322、
1323、在溶于dcm(500ml)内的5-氯-2-(甲硫基)嘧啶-4-羧酸甲酯(47.4g,216.78mmol,1当量)中,分批加入m-cpba(123.23g,606.98mmol,2.8当量,85%纯度)。混合物在21℃下搅拌5小时,获得白色悬浮液。lcms表明起始物质已完全消耗且检测到一个具有所需物质的主峰。两项反应并行进行,随后合于一起,以供后处理。反应物倒入冰水中,并以碳酸钠小心碱化至ph=9~10,随后以dcm萃取(400ml×3)。合并有机层经浓碳酸氢钠水溶液(300ml×5)和卤水洗涤、硫酸镁干燥及过滤后,浓缩至干态。残余物经乙酸乙酯/石油醚=2/1(200ml)研磨、过滤及空气内干燥,获得白色固体的5-氯-2-(甲磺酰基)嘧啶-4-羧酸甲酯(81.2g,收率74.71%)。
1324、lc-ms m/z:251.1(m+1)+。
1325、步骤3:5-氯-2-((3-甲氧基-4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯基)氨基)嘧啶-4-羧酸甲酯
1326、
1327、在溶于dmf(60ml)内的n-(3-甲氧基-4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯基)甲酰胺(6g,18.05mmol,1当量)中,加入60%的氢化钠(1.44g,36.1mmol,2当量)。40℃下搅拌1小时后,在反应物中加入5-氯-2-(甲磺酰基)嘧啶-4-羧酸甲酯(6.79g,27.07mmol,1.5当量)。所得混合物在22℃下搅拌16小时,获得棕色溶液。lcms表明起始物质已完全消耗且检测到一个新的主峰。反应物以水稀释,并以dcm/异丙醇=3/1(80ml×3)萃取。合并有机层以水洗涤(50ml×3)后,有机层以1n盐酸水溶液酸化至ph=2~3,并以dcm洗涤(20ml×3)。水层以碳酸氢钠水溶液碱化至ph=9~10,并以dcm/异丙醇=3/1(80ml×3)萃取。合并有机层经卤水洗涤、硫酸钠干燥、过滤及浓缩,获得棕色胶状物的5-氯-2-((3-甲氧基-4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯基)氨基)嘧啶-4-羧酸甲酯(5.45g,收率63.59%)。
1328、lc-ms m/z:475.2(m+1)+。
1329、步骤4:5-氯-2-((3-甲氧基-4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯基)氨基)嘧啶-4-羧酸
1330、
1331、在溶于甲醇(50ml)内的5-氯-2-((3-甲氧基-4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯基)氨基)嘧啶-4-羧酸甲酯(5.45g,11.47mmol,1当量)中,逐滴加入溶于水(20ml)中的氢氧化钠(0.55g,13.77mmol,1.2当量)。反应物在22℃下搅拌16小时,获得棕色溶液。lcms表明起始物质已完全消耗且检测到一个具有所需物质的主峰。反应物浓缩至干态后,残余物溶于dcm/甲醇=10/1(100ml)。过滤后,滤过物浓缩至干态。所得产物经冻干,获得浅棕色固体的5-氯-2-((3-甲氧基-4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯基)氨基)嘧啶-4-羧酸(4.9g,收率92.63%)。
1332、lc-ms m/z:461.2(m+1)+。
1333、步骤5:5-氯-n-(2-氰基-6-甲氧基苯基)-2-((3-甲氧基-4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺
1334、
1335、0℃下,在溶于吡啶(2ml)内的5-氯-2-((3-甲氧基-4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯基)氨基)嘧啶-4-羧酸(150mg,325.41μmol,1当量)及2-氨基-3-甲氧基苯腈(57.86mg,390.49μmol,1.2当量)中,加入三氯氧磷(149.68mg,976.22μmol,3当量)。反应物在0~5℃下搅拌0.5小时,获得棕色溶液。lcms表明起始物质已完全消耗且检测到一个具有所需物质的主峰。反应物以浓碳酸氢钠水溶液碱化至ph=9~10,并以dcm/异丙醇=3/1(10ml×3)萃取。合并有机层经卤水洗涤、硫酸镁干燥及过滤,并浓缩至干态。残余物经快速柱纯化(c18,以含0.1%甲酸的54%乙腈水溶液洗脱),流出物经冻干,获得浅棕色固体的实施例206(59.14mg,收率30.75%)。
1336、lc-ms m/z:591.2(m+1)+。
1337、1h nmr(400mhz,甲醇-d4)δ8.59(s,1h),7.57~7.37(m,4h),7.27(dd,j=8.7,2.4hz,1h),6.97(d,j=8.6hz,1h),3.91(d,j=21.8hz,6h),3.38(d,j=12.4hz,2h),3.09(s,4h),2.84(s,4h),2.77(t,j=11.8hz,2h),2.70(s,3h),2.66~2.54(m,1h),2.13(d,j=13.4hz,2h),1.78(q,j=12.8,12.0hz,2h)。
1338、
1339、
1340、实施例209:
1341、5-氯-n-(2-氯-6-(三氟甲基)苯基)-2-((4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺
1342、
1343、步骤1:1-(1-甲基哌啶-4-基)-4-(4-硝基苯基)哌嗪
1344、
1345、在溶于二氯乙烯(100ml)内的1-(4-硝基苯基)哌嗪(5g,24.13mmol,1当量)中,加入1-甲基哌啶-4-酮(2.87g,25.33mmol,1.05当量)。18℃下搅拌1小时后,反应物中加入三乙酰氧基硼氢化钠(10.23g,48.25mmol,2当量)。混合物在20℃下搅拌16小时,获得黄色悬浮液。lcms表明起始物质已完全消耗且检测到一个具有所需物质的峰。反应物以浓碳酸氢钠水溶液碱化至ph=9~10,并以dcm/异丙醇=3/1(100ml×3)萃取。合并有机层经卤水洗涤、硫酸镁干燥、过滤及浓缩,获得黄色固体的1-(1-甲基哌啶-4-基)-4-(4-硝基苯基)哌嗪(7.1g,收率96.73%)。
1346、lc-ms m/z:305.2(m+1)+。
1347、步骤2:4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯胺
1348、
1349、氮气气氛中,在溶于甲醇(100ml)内的1-(1-甲基哌啶-4-基)-4-(4-硝基苯基)哌嗪(7.1g,23.33mmol,1当量)中,加入钯碳(0.6g)。反应物以氢气脱气数次,并在20℃下搅拌16小时,获得黑棕色悬浮液。lcms表明起始物质已完全消耗且检测到一个具有所需物质的主峰。反应物以硅藻土垫过滤后,滤饼以甲醇洗涤。滤过物经浓缩获得红色固体的4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯胺(6.6g,收率103.13%)。
1350、lc-ms m/z:275.2(m+1)+。
1351、步骤3:n-(4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯基)甲酰胺
1352、
1353、将溶于甲酸(30ml)内的4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯胺(5.6g,20.41mmol,1当量)在80℃下搅拌3小时,获得紫色溶液。lcms表明起始物质已完全消耗和检测到具有所需物质的一个新峰。反应物以浓碳酸钠水溶液碱化至ph=10~11,并以dcm/异丙醇=3/1(60ml×3)萃取。合并有机层经卤水洗涤、硫酸镁干燥、过滤及浓缩,获得紫色固体的n-(4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯基)甲酰胺(5.5g,收率89.14%)。
1354、lc-ms m/z:303.2(m+1)+。
1355、步骤4:5-氯-2-((4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯基)氨基)嘧啶-4-羧酸
1356、
1357、在溶于dmf(30ml)内的n-(4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯基)甲酰胺(2.5g,8.27mmol,1当量)中,加入60%的氢化钠(0.66g,16.53mmol,2当量)。40℃下搅拌1小时后,反应物中加入5-氯-2-(甲磺酰基)嘧啶-4-羧酸甲酯(2.07g,8.27mmol,1当量)。所得混合物在21℃下搅拌16小时,获得棕色溶液。lcms表明起始物质剩下大约一半且检测到一个新的峰。反应物中加入溶于水(20ml)中的氢氧化钠(1g,24.8mmol,3当量),并继续搅拌4小时,获得棕色溶液。lcms表明检测到一个具有所需物质的主峰。反应物以水稀释,并以乙酸乙酯洗涤(20ml)。水层以1n盐酸水溶液酸化至ph=2~3,并以dcm洗涤(20ml×3)。水层浓缩至干态后,将残余物溶于dcm/甲醇=10/1(50ml)中,并进行过滤。滤过物经浓缩获得棕色固体的5-氯-2-((4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯基)氨基)嘧啶-4-羧酸(1g,收率28.09%)。
1358、lc-ms m/z:431.2(m+1)+。
1359、步骤5:5-氯-n-(2-氯-6-(三氟甲基)苯基)-2-((4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯基)氨基)嘧啶-4-羧酰胺
1360、
1361、0℃下,在溶于吡啶(2ml)内的5-氯-2-((4-(4-(1-甲基哌啶-4-基)哌嗪-1-基)苯基)氨基)嘧啶-4-羧酸(150mg,348.08μmol,1当量)及2-氯-6-(三氟甲基)苯胺(136.15mg,696.16μmol,2当量)中,加入三氯氧磷(266.84mg,1.74mmol,5当量)。反应物在21℃下搅拌0.5小时,获得棕色溶液。lcms表明起始物质已完全消耗且检测到一个具有所需物质的峰。反应物以浓碳酸氢钠水溶液碱化至ph=9~10,并以dcm/异丙醇=3/1(10ml×3)萃取。合并有机层经卤水洗涤、硫酸镁干燥及过滤后,浓缩至干态。残余物经快速柱纯化(c18,以含0.1%甲酸的52%乙腈水溶液洗脱)后,流出物经冻干,获得浅棕色固体的实施例209(27.19mg,收率12.84%)。
1362、lc-ms m/z:608.2(m+1)+;
1363、1h nmr(400mhz,甲醇-d4)δ8.56(s,1h),7.90(dd,j=8.2,1.4hz,1h),7.81(dd,j=8.0,1.4hz,1h),7.66(s,1h),
1364、7.66~7.57(m,2h),7.04~6.95(m,2h),3.45~3.37(m,2h),3.20(t,j=5.0hz,4h),2.83(t,j=5.0hz,6h),2.73(s,3h),2.57(d,j=11.4hz,1h),2.20~2.12(m,2h),1.79(s,2h)。
1365、
1366、
1367、
1368、
1369、实施例215:
1370、5-氯-n-(2-氯-6-氰基苯基)-2-((4-(4-吗啉代哌啶-1-基)苯基)氨基)嘧啶-4-羧酰胺
1371、
1372、路线图
1373、
1374、步骤1:n-(4-(4-吗啉代哌啶-1-基)苯基)甲酰胺
1375、
1376、将溶于甲酸(20ml)内的4-(4-吗啉代哌啶-1-基)苯胺(5g,19.13mmol,1当量)在100℃下搅拌4小时,获得黑色悬浮液。lcms表明起始物质已完全消耗且检测到一个具有所需物质的主峰。反应混合物冷却至室温后,进行浓缩。水相以碳酸钠水溶液碱化至ph=10,并以dcm萃取。合并有机层经卤水洗涤、硫酸钠干燥、过滤及浓缩,获得紫色油状物的n-(4-(4-吗啉代哌啶-1-基)苯基)甲酰胺(6g,108%)。
1377、lc-ms m/z:289.38(m+1)+。
1378、步骤3:5-氯-2-((4-(4-吗啉代哌啶-1-基)苯基)氨基)嘧啶-4-羧酸
1379、
1380、在溶于dmf(5ml)内的n-(4-(4-吗啉代哌啶-1-基)苯基)甲酰胺(0.5g,1.73mmol,1当量)中,加入氢化钠(0.11g,2.59mmol,1.5当量),并在40℃下搅拌1小时。在该溶液中加入5-氯-2-(甲磺酰基)嘧啶-4-羧酸甲酯(0.48g,1.9mmol,1.1当量)后,继续搅拌16小时。lcms表明起始物质已大部分消耗且检测到一个峰。反应物中加入氢氧化钠(0.5g)及水(3ml)后,继续搅拌2小时。反应物以水稀释,并以乙酸乙酯萃取。水相以1n盐酸水溶液酸化至ph=2~3后,以dcm萃取。合并有机层经卤水洗涤、硫酸钠干燥、过滤及浓缩,获得黄色固体的5-氯-2-((4-(4-吗啉代哌啶-1-基)苯基)氨基)嘧啶-4-羧酸(0.4g,50%)。
1381、lc-ms m/z:417.89(m+1)+。
1382、步骤4:5-氯-n-(2-氯-6-氰基苯基)-2-((3-氟-4-(4-吗啉代哌啶-1-基)苯基)氨基)嘧啶-4-羧酰胺
1383、
1384、0℃下,在溶于吡啶(1ml)中的5-氯-2-((4-(4-吗啉代哌啶-1-基)苯基)氨基)嘧啶-4-羧酸(0.1g,0.25mmol,1当量)中,加入2-氨基-3-氯苯腈(0.04g,0.31mmol,1.2当量),随后加入三氯氧磷(0.06g,0.38mmol,1.5当量),并搅拌0.5小时。lcms表明起始物质已大部分消耗且检测到一个峰。反应物经浓缩后,残余物经c18色谱纯化(0%~100%的乙腈水溶液,combiflash nextgen 300),获得黄色固体的实施例215(21mg,9%,甲酸盐)。
1385、lc-ms m/z:553.45(m+1)+;
1386、1h nmr(400mhz,甲醇-d4)δ8.57(s,1h),7.88(ddd,j=25.3,8.0,1.4hz,2h),7.64(d,j=8.8hz,2h),7.56(t,j=8.0hz,1h),7.09~6.92(m,2h),3.85(t,j=4.7hz,4h),3.80~3.70(m,2h),3.04(t,j=4.8hz,4h),3.01(s,1h),2.91~2.81(m,2h),2.74(td,j=12.4,2.3hz,2h),2.16(dt,j=12.2,3.3hz,2h),1.77(qd,j=12.1,4.0hz,2h)。
1387、
1388、
1389、实施例218:
1390、5-氯-n-(2,6-二氯苯基)-2-((4-(4-吗啉代哌啶-1-基)苯基)氨基)嘧啶-4-羧酰胺
1391、
1392、路线图
1393、
1394、步骤1:n-(4-(4-吗啉代哌啶-1-基)苯基)甲酰胺
1395、
1396、将溶于甲酸(20ml)中的4-(4-吗啉代哌啶-1-基)苯胺(5g,19.13mmol,1当量)在80℃下搅拌4小时,获得黑色悬浮液。lcms表明起始物质已完全消耗且检测到一个具有所需物质的主峰。反应混合物冷却至室温,并进行浓缩。水相以碳酸钠水溶液碱化至ph=10,并以dcm/甲醇(3/1)萃取。合并有机层以卤水洗涤、硫酸钠干燥、过滤及浓缩,获得紫色固体的n-(4-(4-吗啉代哌啶-1-基)苯基)甲酰胺(6g,108%)。
1397、lc-ms m/z:290.78(m+1)+。
1398、步骤2:5-氯-n-(2,6-二氯苯基)-2-((4-(4-吗啉代哌啶-1-基)苯基)氨基)嘧啶-4-羧酰胺
1399、
1400、在溶于dmf(2ml)内的n-(4-(4-吗啉代哌啶-1-基)苯基)甲酰胺(83.63mg,0.29mmol,1.1当量)中,加入氢化钠(15.8mg,0.39mmol,1.5当量),并在40℃下搅拌1小时。在溶液中加入5-氯-n-(2,6-二氯苯基)-2-(甲磺酰基)嘧啶-4-羧酰胺(100mg,0.26mmol,1当量)后,搅拌16小时,获得棕色悬浮液。lcms表明起始物质已大部分消耗且检测到一个峰。反应物中加入氢氧化钠水溶液(0.5ml)后,搅拌2小时。反应物以水稀释,并以乙酸乙酯萃取。合并有机层经卤水洗涤、硫酸钠干燥、过滤及浓缩,残余物经c18色谱纯化(0%~100%的乙腈水溶液,combiflash nextgen 300),获得橙色固体的实施例218(88mg,61%,甲酸盐)。
1401、lc-ms m/z:561.13(m+1)+;
1402、1h nmr(400mhz,甲醇-d4)δ8.55(s,1h),7.71~7.60(m,2h),7.55(d,j=8.1hz,2h),7.39(dd,j=8.7,7.6hz,1h),7.14~6.89(m,2h),3.95(s,4h),3.81(dq,j=12.5,2.3hz,2h),2.78(td,j=12.5,2.2hz,2h),2.25(dt,j=10.4,3.2hz,2h),1.86(qd,j=12.1,4.1hz,2h)。
1403、
1404、
1405、实施例221
1406、5-氯-n-(2-氰基-6-甲氧基苯基)-2-((3-氟-4-(4-甲基-4,7-二氮杂螺[2.5]辛烷-7-基)苯基)氨基)嘧啶-4-羧酰胺
1407、
1408、路线图
1409、
1410、步骤1:3-氟-4-(4-甲基-4,7-二氮杂螺[2.5]辛烷-7-基)苯胺
1411、
1412、在溶于乙醇(10ml)内的7-(2-氟-4-硝基苯基)-4-甲基-4,7-二氮杂螺[2.5]辛烷(0.7g,2.64mmol,1当量)中,加入二水合氯化亚锡(2.38g,10.55mmol,4当量),并在70℃下搅拌4小时,获得红色悬浮液。lcms表明起始物质已完全消耗且检测到一个具有所需物质的主峰。反应混合物冷却至室温后,进行浓缩。水相以碳酸钠水溶液碱化至ph=10,且以dcm萃取。合并有机层经卤水洗涤、硫酸钠干燥、过滤及浓缩,获得橙色油状物的3-氟-4-(4-甲基-4,7-二氮杂螺[2.5]辛烷-7-基)苯胺(0.5g,80%)。
1413、lc-ms m/z:236.15(m+1)+。
1414、步骤2:n-(3-氟-4-(4-甲基-4,7-二氮杂螺[2.5]辛烷-7-基)苯基)甲酰胺
1415、
1416、将溶于甲酸(3ml)内的3-氟-4-(4-甲基-4,7-二氮杂螺[2.5]辛烷-7-基)苯胺(0.3g,1.27mmol,1当量)在100℃下搅拌4小时,获得黑色悬浮液。lcms表明起始物质已完全消耗且检测到一个具有所需物质的主峰。反应混合物冷却至室温后,进行浓缩。水相以碳酸钠水溶液碱化至ph=10,并以dcm萃取。合并有机层经卤水洗涤、硫酸钠干燥、过滤及浓缩,获得棕色油状物的n-(3-氟-4-(4-甲基-4,7-二氮杂螺[2.5]辛烷-7-基)苯基)甲酰胺(0.5g,148%)。
1417、lc-ms m/z:264.14(m+1)+。
1418、步骤3:5-氯-2-((3-氟-4-(4-甲基-4,7-二氮杂螺[2.5]辛烷-7-基)苯基)氨基)嘧啶-4-羧酸
1419、
1420、在溶于dmf(5ml)中的n-(3-氟-4-(4-甲基-4,7-二氮杂螺[2.5]辛烷-7-基)苯基)甲酰胺(0.5g,1.90mmol,1当量)中,加入氢化钠(0.15g,3.80mmol,2当量),并在40℃下搅拌1小时。在溶液中加入5-氯-2-(甲磺酰基)嘧啶-4-羧酸甲酯(0.52g,2.09mmol,1.1当量)后,搅拌16小时。lcms表明起始物质已大部分消耗且检测到一个峰。反应物中加入氢氧化钠(0.5g)及水(3ml)后,搅拌2小时。反应物以水稀释,并以乙酸乙酯萃取。水相以1n盐酸水溶液酸化至ph=2~3,并以dcm萃取。合并有机层经卤水洗涤、硫酸钠干燥、过滤及浓缩,获得黄色固体的5-氯-2-((3-氟-4-(4-甲基-4,7-二氮杂螺[2.5]辛烷-7-基)苯基)氨基)嘧啶-4-羧酸(0.1g,12%)。
1421、lc-ms m/z:392.12(m+1)+。
1422、步骤4:5-氯-n-(2-氰基-6-甲氧基苯基)-2-((3-氟-4-(4-甲基-4,7-二氮杂螺[2.5]辛烷-7-基)苯基)氨基)嘧啶-4-羧酰胺
1423、
1424、0℃下,在溶于吡啶(1ml)内的5-氯-2-((3-氟-4-(4-甲基-4,7-二氮杂螺[2.5]辛烷-7-基)苯基)氨基)嘧啶-4-羧酸(0.1g,0.25mmol,1当量)中,加入2-氨基-3-甲氧基苯腈(0.04g,0.31mmol,1.2当量),然后加入三氯氧磷(0.06g,0.38mmol,1.5当量),并搅拌0.5小时。lcms表明起始物质已大部分消耗且检测到一个峰。反应物浓缩后,残余物经c18色谱纯化(0%~100%的乙腈水溶液,combiflash nextgen 300),获得黄色固体的实施例221(9mg,7%,甲酸盐)。
1425、lc-ms m/z:522.17(m+1)+;
1426、1h nmr(400mhz,甲醇-d4)δ8.62(s,1h),7.69(dd,j=14.9,2.5hz,1h),7.59~7.34(m,4h),7.07(t,j=9.2hz,1h),3.96(s,3h),3.24(s,4h),3.01(s,2h),2.60(s,3h),1.05~0.67(m,4h)。
1427、
1428、
1429、实施例224
1430、n-(5-氯-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)-4-甲基烟酰胺
1431、
1432、路线图
1433、
1434、步骤1:4-甲基烟酰氯
1435、
1436、0℃下,在溶于dcm(30ml)内的4-甲基烟酸化合物(1g,7.29mmol,1当量)中,加入乙二酰氯(4.63g,36.46mmol,5当量)及dmf(53.30mg,0.73mmol,0.1当量)。反应物室温搅拌1小时后,获得黄色溶液。tlc(以甲醇淬灭)表明起始物质已完全消耗且检测到一个新的斑点。反应物浓缩至干态后,残余物以dcm进行交换(20ml×3),获得黄色固体的4-甲基烟酰氯(1.14g,收率:100.9%)。
1437、步骤2:n-(2,5-二氯嘧啶-4-基)-4-甲基烟酰胺
1438、
1439、0℃下,在混于thf(10ml)内的2,5-二氯嘧啶-4-胺(1g,6.10mmol,1当量)中,加入氢化钠(0.37g,9.15mmol,1.5当量)。搅拌0.5小时后,0℃下,在反应物中逐滴加入溶于thf(5ml)和dcm(5ml)中的4-甲基烟酰氯。反应物室温搅拌16小时,获得棕色溶液。反应混合物的lcms分析表明完全转化至所需产物。反应以冰水淬灭后,反应物以0.5n盐酸酸化至ph=2~3,并以乙酸乙酯洗涤(10ml×3)。水层以浓碳酸钠水溶液碱化至ph=10~11,且以dcm萃取(20ml×3)。合并有机层经卤水洗涤、硫酸钠干燥及过滤后,浓缩至干态。残余物经硅胶纯化(石油醚:乙酸乙酯=0:60),获得米白色固体的n-(2,5-二氯嘧啶-4-基)-4-甲基烟酰胺(0.41g,收率:39.42%)。
1440、lc-ms m/z:283.01(m+1)+。
1441、步骤3:n-(5-氯-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)-4-甲基烟酰胺
1442、
1443、将溶于异丙醇(1.5ml)内的n-(2,5-二氯嘧啶-4-基)-4-甲基烟酰胺化合物(50mg,0.177mmol,1当量)及4-(4-甲基哌嗪-1-基)苯胺(37.16mg,0.194mmol,1.2当量)100℃下搅拌1小时。lcms分析表明检测到所需物质且反应完成。冷却至室温后,混合物进行过滤,随后小心倒入10ml的水和二氯甲烷中。通过加入盐酸调至ph=5~6后,分离水相。水相以氢氧化钠水溶液碱化至ph=7~8,并以二氯甲烷萃取。有机层经浓缩和制备型tlc纯化,获得淡黄色固体的实施例224。
1444、lc-ms m/z:438.1(m+1)+;
1445、1h nmr(400mhz,d6-dmso):δ9.63(s,1h),8.61(s,1h),8.53(d,j=5.0hz,1h),8.46(s,1h),7.51(d,j=8.7hz,2h),7.36(d,j=5.3hz,1h),6.86(d,j=8.5hz,2h),3.12(s,3h),2.80(s,3h),2.43(s,4h),1.20(s,4h)。
1446、按照实施例67的方法,合成以下化合物。
1447、
1448、实施例226:
1449、2-氟-n-(5-氟-2-((2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)苯甲酰胺
1450、
1451、路线图
1452、
1453、步骤1:二叔丁氧羰基-5-氯-2-氟嘧啶-4-胺
1454、
1455、在溶于dcm(20ml)内的5-氯-2-氟嘧啶-4-胺(2g,13.56mmol,1当量)中,加入二碳酸二叔丁酯(11.83g,54.22mmol,4当量)、三乙胺(1.37g,13.56mmol,1当量)及4-二甲氨基吡啶(0.16g,1.36mmol,0.1当量)。反应物在25℃下搅拌16小时,获得黄色溶液。tlc1表明起始物质已完全消耗且检测到一个新的斑点。反应物以水稀释,并以dcm萃取。合并有机层经卤水洗涤、硫酸钠干燥及过滤后,浓缩至干态。残余物经快速柱纯化(二氧化硅,溶有0%~20%乙酸乙酯的石油醚)后,收集流出物,并将其冻干,获得白色固体的二叔丁氧羰基-5-氯-2-氟嘧啶-4-胺(3.4g,70%)。
1456、lc-ms m/z:348.1(m+1)+。
1457、步骤2:二叔丁氧羰基-5-氟-n2-(2-甲基-1,2,3,4-四氢异喹啉-7-基)嘧啶-2,4-二胺
1458、
1459、氮气气氛中,在溶于二恶烷(1ml)内的二叔丁氧羰基-5-氯-2-氟嘧啶-4-胺(100mg,287.55μmol,1当量)及2-甲基-1,2,3,4-四氢异喹啉-7-胺(46.65mg,287.55μmol,1当量)中,加入xantphos(32.3mg,57.51μmol,0.2当量)、碳酸铯(187.38mg,575.09μmol,2当量)及乙酸钯(6.46mg,28.75μmol,0.1当量)。所得混合物在100℃下微波搅拌40分钟,获得棕色悬浮液。lcms(y0029-42-p1a1)表明起始物质已完全消耗且检测到一个具有所需物质的主峰。反应混合物冷却至室温后,经过滤和浓缩,获得二叔丁氧羰基-5-氟-n2-(2-甲基-1,2,3,4-四氢异喹啉-7-基)嘧啶-2,4-二胺。
1460、lc-ms m/z:474.24(m+1)+。
1461、步骤3:5-氟-n2-(2-甲基-1,2,3,4-四氢异喹啉-7-基)嘧啶-2,4-二胺
1462、
1463、在溶于dcm(1ml)内的二叔丁氧羰基-5-氟-n2-(2-甲基-1,2,3,4-四氢异喹啉-7-基)嘧啶-2,4-二胺(287.55μmol,1当量)中,加入盐酸二恶烷(2ml)。所得混合物搅拌1小时后,获得黄色悬浮液。lcms表明起始物质已完全消耗且检测到一个具有所需物质的主峰。反应混合物经浓缩,获得5-氟-n2-(2-甲基-1,2,3,4-四氢异喹啉-7-基)嘧啶-2,4-二胺(0.19g,241%)。
1464、lc-ms m/z:274.24(m+1)+。
1465、步骤4:2-氟-n-(5-氟-2-((2-甲基-1,2,3,4-四氢异喹啉-7-基)氨基)嘧啶-4-基)苯甲酰胺
1466、
1467、0℃下,在溶于dcm(1ml)内的2-氟苯甲酸(50mg,0.35mmol,1当量)中,加入乙二酰氯(226mg,1.78mmol,5当量)及dmf。反应物在25℃下搅拌1小时,获得淡黄色溶液。反应物浓缩至干态后,残余物以dcm进行交换(10ml×3),获得浅黄色胶状物。0℃下,在溶于dmf(1ml)内的5-氟-n2-(2-甲基-1,2,3,4-四氢异喹啉-7-基)嘧啶-2,4-二胺(97mg,0.35mmol,1当量)中,加入氢化钠(71mg,1.78mmol,5当量)。搅拌1小时后,0℃下,在反应物中逐滴加入溶于dmf(1ml)内的sm1(50mg,0.35mmol,1当量)。反应物在25℃下搅拌16小时,获得黄色悬浮液。lcms表明起始物质存在残留且检测到一个主峰。反应物以水稀释,并以乙酸乙酯萃取。合并有机层经氯化铵水溶液和卤水洗涤、硫酸钠干燥、过滤及浓缩,残余物经c18色谱纯化(0%~100%的乙腈水溶液,combiflash nextgen 300),获得白色固体的实施例226(1.4mg,2.8%,甲酸盐)。
1468、lc-ms m/z:396.06(m+1)+;
1469、1h nmr(400mhz,甲醇-d4)δ8.36(d,j=2.9hz,1h),8.03(d,j=2.3hz,1h),7.86(td,j=7.5,1.8hz,1h),7.65(dddd,j=8.5,7.2,5.2,1.8hz,1h),7.46(dd,j=8.4,2.4hz,1h),7.41~7.26(m,2h),7.20(d,j=8.4hz,1h),4.57(d,j=15.2hz,1h),4.36(d,j=15.2hz,1h),3.76(s,1h),3.43(t,j=12.4hz,1h),3.29~3.11(m,2h),3.08(s,3h)。
1470、
1471、
1472、实施例230:
1473、2-氟-6-羟基-n-(2-((6-甲基-5,6,7,8-四氢-1,6-萘啶-3-基)氨基)-5-乙烯基嘧啶-4-基)苯甲酰胺
1474、
1475、路线图
1476、
1477、步骤1:2-氯-5-乙烯基嘧啶-4-胺
1478、
1479、氮气气氛中,在溶于dmf(10ml)内的5-溴-2-氯嘧啶-4-胺(1g,4.8mmol,1当量)中,加入三(2-呋喃基)膦(0.22g,0.96mmol,0.2当量)及pd2(dba)3(0.13g,0.14mmol,0.03当量)。25℃下搅拌10分钟后,在反应物中加入三丁基乙烯基锡(1.98g,6.24mmol,1.3当量)。反应物在65℃下微波搅拌0.5小时,获得淡黄色溶液。lcms表明,大部分起始物质残留,且检测到具有所需物质的一个新峰。反应物在70℃下继续微波搅拌1小时,获得浅棕色溶液。lcms表明反应完成。反应物以乙酸乙酯稀释,并以浓氟化鉀水溶液淬灭。混合物以硅藻土垫过滤,并以乙酸乙酯萃取(20ml×3)。合并有机层经卤水洗涤、硫酸钠干燥及过滤后,浓缩至干态。残余物经快速柱纯化(二氧化硅,以溶有15%乙酸乙酯的石油醚洗脱,plate 1),获得淡黄色固体的2-氯-5-乙烯基嘧啶-4-胺(0.63g,收率84.4%)。
1480、步骤2:2-(烯丙氧基)-6-氟苯甲酸
1481、
1482、在溶于乙腈(15ml)内的2-氟-6-羟基苯甲酸(1.5g,9.61mmol,1当量)中,加入碳酸钾(3.32g,24.02mmol,2.5当量)及3-溴-1-丙烯(2.91g,24.02mmol,2.5当量)。反应物在80℃下搅拌3小时,获得粉红色悬浮液。lcms表明反应起始物质已完全消耗且检测到一个新的主峰。反应物以水稀释,并以1n盐酸水溶液酸化至ph=5。混合物以dcm萃取(30ml×3)后,合并有机层经卤水洗涤、硫酸钠干燥及过滤,并浓缩至干态。残余物溶于甲醇(15ml)中,并加入溶于水(5ml)中的氢氧化钠(0.42g,10.57mmol,1.1当量)。反应物在80℃下搅拌2小时,获得浅棕色溶液。lcms表明检测到一个具有所需物质的主峰。反应物以水稀释,并以1n盐酸水溶液酸化至ph=2。混合物以dcm萃取(30ml×3)后,合并有机层经卤水洗涤、硫酸钠干燥、过滤及浓缩,获得浅棕色油状物的2-(烯丙氧基)-6-氟苯甲酸(1.56g,收率82.98%)。
1483、lc-ms m/z:197.1(m+1)+。
1484、步骤3:2-(烯丙氧基)-n-(2-氯-5-乙烯基嘧啶-4-基)-6-氟苯甲酰胺
1485、
1486、0℃下,在氮气气氛中,将乙二酰氯(2.57g,20.25mmol,5当量)和一滴dmf加入溶于dcm(10ml)内的2-(烯丙氧基)-6-氟苯甲酸(1.03g,5.26mmol,1.3当量)中。25℃下搅拌30分钟后,反应物浓缩至干态。0℃下,在氮气气氛中,将氢化钠(0.32g,8.1mmol,2当量)加入溶于dmf(10ml)内的2-氯-5-乙烯基嘧啶-4-胺(0.63g,4.05mmol,1当量)中。25℃下搅拌30分钟后,加入溶于dcm(10ml)内的苯甲酰氯。反应物在25℃下搅拌16小时,获得浅棕色溶液。lcms表明大部分起始物质已消耗且检测到具有所需物质的一个新峰。反应以氯化铵水溶液淬灭,并以乙酸乙酯萃取(20ml×3)。合并有机层经卤水洗涤、硫酸钠干燥及过滤后,浓缩至干态。残余物经快速柱纯化(二氧化硅,以溶有18%乙酸乙酯的石油醚洗脱,plate 1),获得米白色固体的2-(烯丙氧基)-n-(2-氯-5-乙烯基嘧啶-4-基)-6-氟苯甲酰胺(0.49g,收率40.16%)。
1487、lc-ms m/z:334.1(m+1)+。
1488、步骤4:2-氟-6-羟基-n-(2-((6-甲基-5,6,7,8-四氢-1,6-萘啶-3-基)氨基)-5-乙烯基嘧啶-4-基)苯甲酰胺
1489、
1490、氮气气氛中,在溶于二恶烷(1ml)内的2-(烯丙氧基)-n-(2-氯-5-乙烯基嘧啶-4-基)-6-氟苯甲酰胺(50mg,149.81μmol,1当量)及6-甲基-5,6,7,8-四氢-1,6-萘啶-3-胺(31.79mg,194.76μmol,1.3当量)中,加入xantphos(8.67mg,14.98μmol,0.1当量)、碳酸铯(73.22mg,224.72μmol,1.5当量)及乙酸钯(1.68mg,7.49μmol,0.05当量)。反应物在90℃下微波搅拌0.5小时,获得棕色溶液。lcms表明起始物质已完全消耗且检测到一个具有实施例5物质的新峰。反应物以硅藻土垫过滤,滤过物以水稀释,并以dcm萃取(10ml×3)。合并有机层经卤水洗涤、硫酸钠干燥及过滤后,浓缩至干态。残余物经快速柱纯化(c18,以含0.05%tfa的60%乙腈水溶液洗脱,流速为60ml/min)。流出物收集后以3n的盐酸水溶液(2滴)冻干,获得淡黄色固体的实施例5的实施例230(1.1mg,收率1.61%)。
1491、lc-ms m/z:421.2(m+1)+;
1492、1h nmr(400mhz,甲醇-d4)δ8.84(q,j=2.7hz,2h),8.69~8.64(m,1h),7.40(td,j=8.4,6.6hz,1h),6.86~6.76(m,2h),6.79~6.71(m,1h),5.80(dd,j=17.5,1.0hz,1h),5.41(dd,j=11.1,1.0hz,1h),4.59(s,2h),3.74(s,2h),3.29(d,j=6.5hz,2h),3.13(s,3h)。
1493、实施例231:
1494、5-氯-n-(2-氰基-6-甲氧基苯基)-2-((3-氟-4-(6-(2-羟基乙基)-3,6-二氮杂二环[3.1.1]庚烷-3-基)苯基)氨基)嘧啶-4-羧酰胺
1495、
1496、步骤:5-氯-n-(2-氰基-6-甲氧基苯基)-2-((3-氟-4-(6-(2-羟基乙基)-3,6-二氮杂二环[3.1.1]庚烷-3-基)苯基)氨基)嘧啶-4-羧酰胺
1497、
1498、在溶于dmf(2ml)中的2-((4-(3,6-二氮杂二环[3.1.1]庚烷-3-基)-3-氟苯基)氨基)-5-氯-n-(2-氰基-6-甲氧基苯基)嘧啶-4-羧酰胺(100mg,202.46μmol,1当量)及2-碘乙-1-醇(52.22mg,303.69μmol,1.5当量)中,加入碳酸钾(83.94mg,607.38μmol,3当量)。反应物在100℃下搅拌1小时,获得棕色溶液。lcms表明起始物质已完全消耗且检测到一个具有所需物质的峰。反应物经快速柱纯化(c18,以含1%甲酸的55%乙腈水溶液洗脱)后,将流出物冻干,获得黄色固体的实施例8的实施例231化合物(10.7mg,收率9.05%)。
1499、lc-ms m/z:538.2(m+1)+;
1500、1h nmr(400mhz,甲醇-d4)δ8.57(d,j=17.5hz,2h),7.68(dd,j=16.6,2.5hz,1h),7.55~7.43(m,2h),7.47~7.37(m,2h),7.06(t,j=9.4hz,1h),4.31(s,2h),3.97(d,j=12.5hz,1h),3.97(s,3h),3.83(s,4h),3.00(s,4h),2.20(s,1h)。
1501、hpk1酶法分析
1502、制备待测化合物的10mm dmso溶液,作为母液。通过以hpk1激酶分析缓冲液进行5倍10点连续稀释,制备化合物的稀释液。hpk1激酶分析缓冲液含40mm tris(ph=7.5)、20mm氯化镁、0.1mg/ml牛血清白蛋白(bsa)及50μm二硫苏糖醇(dtt)。反应体系内加入7.5ng/μl活性hpk1(promega,v4098)及0.1μg/ml作为底物蛋白的髓鞘碱性蛋白(mbp)。hpk1激酶反应体系内的三磷酸腺苷(atp)最终浓度为10μm。连续稀释化合物转移至反应体系内后,室温温育1小时。加入5μl的adp-glotm试剂(promega,v9101)后,温育40分钟,以终止激酶反应,并耗光未消耗的atp。之后,加入10μl的激酶检测试剂,并室温温育30~60分钟。以读板光度计测量发光量。
1503、以上述实施例为例,本公开化合物的hpk1 ic50(nm)处于如下范围:a:≤100nm;b:100nm<ic50≤1000nm;c:>1000nm。
1504、
1505、
1506、
- 还没有人留言评论。精彩留言会获得点赞!