一种检测结构变异的方法及装置与流程
1.本发明涉及生物信息学领域,具体涉及一种检测结构变异的方法及装置。
背景技术:
2.基因组结构变异(structure variations,sv)是指在基因组上发生较大长度(大于等于50bp)的序列变化和位置变化,包括插入(insertion)、缺失(deletion)、重复(duplication)、倒位(inversion)、易位(translocation)等几种类型,是基因组发生变异的重要来源,与进化、遗传病、肿瘤等有关系。
3.二代测序技术常用的是双端(pair end,pe)测序类型,即对同一模板链(template)正反向各测固定的长度,对应产出数据的两条序列(reads),分别是read1和read2(互为mat e),这些原始序列经过比对软件分析,可以将其在参考基因组上的位置标识出来,正常情况下是匹配良好的,两条序列的比对位置差不会超过模板链长度分布,但存在一些比对,比如:序列分几部分比对到参考基因组的不同位置(切分比对,split reads mapping,sr)、序列末端比对到参考基因组的其他位置或未比对到参考基因组的任何位置(软剪切,soft clip,sc)、两条序列中的一条正常比对另一条未比对上(单条比对,single unmapped,su)、两条序列比对到两个染色体或比对位置差很远(异常对,discordant pair,dp)、比对处的序列覆盖深度异常(覆盖度,depth of coverage,doc)等,这些比对情况可作为支持结构变异检测的信号。
4.有了这些来源于原始reads比对信息的信号,从此结果出发,如何反推可能的结构变异事件,包含发生的染色体、断点、方向等,是近十五年基于二代测序发展的检测结构变异的理论方法和软件应用要解决的问题,这些软件的差异主要体现在:提取信号的类型、对信号的处理方法、是否组装(assembly,as)等方面。
技术实现要素:
5.根据第一方面,在一实施例中,提供一种检测结构变异的方法,包括:
6.计算步骤,包括计算待测样本测序文库的最大插入片段长度;
7.信号提取步骤,包括从待测样本的测序数据中提取su信号、dp信号、sr信号;
8.信号处理步骤,包括对sr信号、su信号、dp信号进行处理,获得结构变异信息。
9.根据第二方面,在一实施例中,提供一种检测结构变异的装置,包括:
10.计算模块,用于计算待测样本测序文库的最大插入片段长度;
11.信号提取模块,用于从待测样本的测序数据中提取su信号、dp信号、sr信号;
12.信号处理模块,用于对sr信号、su信号、dp信号进行处理,获得结构变异信息。
13.根据第三方面,在一实施例中,提供一种装置,包括:
14.存储器,用于存储程序;
15.处理器,用于通过执行所述存储器存储的程序以实现第一方面的方法。
16.根据第四方面,在一实施例中,提供一种计算机可读存储介质,所述介质上存储有
程序,所述程序能够被处理器执行以实现第一方面的方法。
17.依据上述实施例的一种检测结构变异的方法及装置,通过对三种信号的提取及处理,可支持肿瘤基因组结构变异的检测。
附图说明
18.图1为实施例1的结构变异信息检测流程图。
具体实施方式
19.下面通过具体实施方式对本发明作进一步详细说明。其中不同实施方式中类似元件采用了相关联的类似的元件标号。在以下的实施方式中,很多细节描述是为了使得本技术能被更好的理解。然而,本领域技术人员可以毫不费力的认识到,其中部分特征在不同情况下是可以省略的,或者可以由其他元件、材料、方法所替代。在某些情况下,本技术相关的一些操作并没有在说明书中显示或者描述,这是为了避免本技术的核心部分被过多的描述所淹没,而对于本领域技术人员而言,详细描述这些相关操作并不是必要的,他们根据说明书中的描述以及本领域的一般技术知识即可完整了解相关操作。
20.另外,说明书中所描述的特点、操作或者特征可以以任意适当的方式结合形成各种实施方式。同时,方法描述中的各步骤或者动作也可以按照本领域技术人员所能显而易见的方式进行顺序调换或调整。因此,说明书中的各种顺序只是为了清楚描述某一个实施例,并不意味着是必须的顺序,除非另有说明其中某个顺序是必须遵循的。
21.本文中为部件所编序号本身,例如“第一”、“第二”等,仅用于区分所描述的对象,不具有任何顺序或技术含义。
22.术语解释
23.如本文所用,结构变异(structure variations,sv)是指在基因组上发生较大长度(大于等于50bp)的序列变化和位置变化,包括插入(insertion)、缺失(deletion)、重复(dupli cation)、倒位(inversion)、易位(translocation)等几种类型,是基因组发生变异的重要来源,与进化、遗传病、肿瘤等有关系。
24.根据第一方面,在一实施例中,提供一种检测结构变异的方法,包括:
25.计算步骤,包括计算最大插入片段长度;
26.信号提取步骤,包括从待测样本的测序数据中提取su信号(single unmapped,su,单条比对信息)、dp信号(discordant pair,dp,异常对信息)、sr信号(split reads mappi ng,切分比对信息);
27.信号处理步骤,包括对sr信号、su信号、dp信号进行处理,获得结构变异信息。
28.在一实施例中,最大插入片段长度的计算方法如下:包括对符合正态分布的插入片段长度计算均值和方差,均值与三倍方差的和记为最大插入片段长度,该统计量是样本特异性的。
29.在一实施例中,信号提取步骤中,所述su信号包含同时满足如下条件的序列对:flag不是次级比对或补充比对;两条序列中的一条为未比对或其mate序列为未比对,这两条序列中的非未比对序列不是重复比对。
30.在一实施例中,信号提取步骤中,提取su信号时,将符合条件的序列对中flag为
unm ap的序列写出到临时文件并排序,其中,该序列的名字用其mate序列的信息命名,该信息包含比对上的参考基因组的名字、开始位置、终止位置、比对质量。
31.在一实施例中,信号提取步骤中,所述dp信号包含同时满足如下条件的序列对:flag不是secondary、supplementary、dup、umap或munmap,两条序列比对上两个不同的染色体或两条序列对应的模板链长度大于最大插入片段长度。
32.在一实施例中,信号提取步骤中,提取dp信号时,将符合条件的序列对写出到临时文件并排序,其中,序列的名字用其mate序列的信息命名,该信息包含比对上的参考基因组的名字、开始位置、终止位置、比对质量。
33.在一实施例中,信号提取步骤中,所述sr信号包含不满足su信号提取条件和dp信号提取条件,且满足如下条件的序列对:cigar的首部或尾部是soft clip类型的序列。
34.在一实施例中,信号提取步骤中,提取sr信号时,将符合条件的序列的soft clip信息写出到临时文本文件,其中,该soft clip信息包含soft clip位于首部或尾部、比对上的参考基因组的名字和比对位置、比对方向、soft clip序列、soft clip碱基质量、序列比对质量。
35.在一实施例中,所述测序数据包含比对到参考基因组的测序数据。
36.在一实施例中,所述参考基因组包含人参考基因组。
37.在一实施例中,所述参考基因组包含hg19(亦称grch37)基因组、hs37d5基因组、b37基因组、hg18基因组、hg17基因组、hg16基因组或hg38基因组的至少一部分。
38.在一实施例中,所述待测样本包含基因组dna。
39.在一实施例中,所述测序数据包含区域捕获测序数据(亦称靶向捕获测序数据)。
40.在一实施例中,sr信号处理时,包括对soft clip信息的处理,具体包括以下步骤:
41.合并步骤,包括合并多条序列上的soft clip信息,合并后的结果定义为soft,作为sr信号,soft名字包含比对上的参考基因组的名字、比对位置、soft位于序列的首部或尾部、比对方向、soft计数、比对质量、soft序号;
42.重比对步骤,包括对合并步骤获得的soft进行重比对,获得重比对信息;
43.结构变异信息提取步骤,根据soft所在序列的信息(前:原始比对)和soft重比对信息(后)两部分,计算两个候选断点的初步信息:soft所在序列的信息记为第一断点,soft重比对信息记为第二断点,根据所述第一断点、第二断点的信息,确定结构变异信息。
44.在一实施例中,合并步骤中,包括构建有序的soft clip信息集合,将符合条件的soft cli p信息作为独立的sr信号。
45.在一实施例中,合并步骤中,按照soft clip信息位于序列的首部或尾部、比对上的参考基因组的位置信息做初步排序,将soft clip位于首部或尾部、比对上的参考基因组的名字、比对位置、比对方向都相同的soft clip作为一簇,构建得到有序的soft clip集合。
46.在一实施例中,所述簇包括正向序列(forward reads)首部的soft clip簇、反向序列(reverse reads)尾部的soft clip簇。
47.在一实施例中,对于正向序列首部的soft clip簇类型,按照soft clip逆序、去重计数、排序后构建新的soft clip集合。
48.在一实施例中,对于反向序列尾部的soft clip簇类型,按照soft clip原序、去重
计数、排序后构建新的soft clip集合。
49.在一实施例中,合并步骤中,构建得到有序的soft clip集合后,将符合如下条件的信息作为独立的sr信号:前后soft clip比较,若soft clip变短或没有前缀关系,则将该soft cli p及其计数作为独立的sr信号;若soft clip变长且符合前缀关系,则推举最长的soft clip及其计数作为独立的sr信号。
50.在一实施例中,获得独立的sr信号后,过滤去除不符合条件的短序列,将所得文件中每条记录定义为所述soft。
51.在一实施例中,所述不符合条件的短序列包含小于20bp的sr信号读段。
52.在一实施例中,重比对步骤中,包括对合并步骤获得的soft进行单端重比对,获得重比对信息。
53.在一实施例中,结构变异信息提取步骤中,根据soft所在序列的信息(原始比对)和soft重比对信息两部分来计算两个候选断点,进而确定结构变异信息。
54.在一实施例中,结构变异信息提取步骤中,从soft重比对信息中提取如下序列:flag不是secondary、supplementary、dup、umap或munmap的比对结果,计算第二断点。
55.在一实施例中,所述两个候选断点的初步信息包括如下信息:断点所在染色体、断点位置、断点soft所在序列的链向(is_reverse)、断点soft所在序列的融合方向(is_gener)、soft重比对的链向(is_reverse)、soft重比对的融合方向(is_gener)。
56.在一实施例中,所述链向、融合方向具体包括如下四种情况:
57.1)正向序列首部来源的soft重比对后,正向比对到第二断点的左侧;
58.2)正向序列首部来源的soft重比对后,反向互补比对到第二断点的右侧;
59.3)反向序列尾部来源的soft重比对后,正向比对到第二断点的右侧;
60.4)反向序列尾部来源的soft重比对后,反向互补比对到第二断点的左侧。
61.在一实施例中,还可以确定结构变异发生的细节,比如由断点所在染色体、断点位置、断点soft比对链向来确定结构变异的类型。
62.在一实施例中,结构变异信息提取步骤中,为寻找dp信号用到的所述两个断点附近区域包括:soft所在侧延伸10bp的区域、soft重比对侧延伸最大插入片段长度-2*测序序列长度的区域。前述两个区域作为查找区间。
63.在一实施例中,结构变异信息提取步骤中,在两个断点附近区域寻找符合条件的dp信号作为补充,在第一断点附近区域寻找符合条件的正常序列,用于计算结构变异频率。
64.在一实施例中,结构变异信息提取步骤中,所述符合条件的dp信号包括:dp类型的两条序列对应的比对染色体和比对位置均与第一断点、第二断点一致,且链向一致,确定dp信号的质量值和支持个数,找到的dp支持在后续dp信号处理时不再考虑,若未找到dp支持且soft的计数小于2,则说明该sr信号太弱,即该sr信号支持证据不足,可过滤去除该sr信号。
65.在一实施例中,结构变异信息提取步骤中,所述符合条件的dp信号要求链向一致判断时,以soft所在序列对应的mate reads链向作为第一断点附近链向。
66.在一实施例中,结构变异信息提取步骤中,为寻找正常序列用到的所述第一断点附近区域包含:第一断点向左延伸最大插入片段长度到断点之间的区域。
67.在一实施例中,结构变异信息提取步骤中,所述符合条件的正常序列包括:flag不
是secondary、supplementary、dup、umap或munmap的比对序列(reads),不计算模板链长度大于最大插入片段长度或比对到不同染色体的序列(reads),根据比对起始位置在查找区间内的序列个数来确定正常序列的支持个数。
68.在一实施例中,sr信号处理时,还包括汇总步骤,具体是在结构变异信息提取步骤后,由于多个soft确定的两个断点可能一样,需要合并为一个。如果多个soft确定的两个断点相同,则合并为一个断点。
69.在一实施例中,合并断点的方法包括:以两个断点所在染色体、断点位置、断点soft所在序列的链向(is_reverse)和融合方向(is_gener)、soft重比对的链向(is_reverse)和融合方向(is_gener)为索引进行合并,并更新sr信号支持个数。
70.在一实施例中,su信号处理时,将提取的su信号重比对后做局部组装,获得结构变异信息。由于组装后回比到参考基因组的序列很少,所以su信号处理后检出的结构变异也很少。
71.在一实施例中,dp信号处理时,dp信号的处理方法包括:正向序列(forward reads)位于可能结构变异断点的左侧(通常是正向序列的大部分位于可能结构变异断点的左侧),反向序列(reverse reads)位于可能结构变异断点的右侧(通常是反向序列的大部分位于可能结构变异断点的右侧),基于这两点规律,区分正向链和反向链,对dp信号的序列(已基于比对起始位置排过序)进行聚簇:相邻序列比较,若比对到相同染色体且比对起始位置相差≤200bp,则完成第一次聚簇,此时确定第一候选断点;对此次结果按照固定次序再次排序(这里比对位置和mate比对位置比较,按照整个基因组水平排序,可能对应mate为多簇,需排序),对mate部分聚簇,相邻序列比较,若比对到相同染色体且比对起始位置相差≤200bp,则完成第二次聚簇,此时确定第二候选断点;两个候选断点附近配对成簇的dp信号得以初步确定。
72.在一实施例中,dp信号的处理方法中,第二候选断点可能不止一个,若前两次聚簇后的序列个数≥5,则进行第三次聚簇,此时如果多个第二候选断点相差》200bp,则分开成簇,再次返回序列个数≥5,获得最终的dp信号。
73.在一实施例中,dp信号的处理方法中,若sr信号处理和su信号处理时已标记过dp信号,该dp信号在dp信号处理时跳过。
74.在一实施例中,dp信号的处理方法中,确定正常序列的支持个数(同sr信号),以多的一侧作为第一断点(breakpoint1)的来源。
75.在一实施例中,dp信号的处理方法中,dp信号的两个断点所在染色体、断点位置、链向、融合方向得以确定后,获得结构变异的类型。
76.在一实施例中,所有信号处理完成后,获得结构变异信息,所述结构变异信息包含如下信息:两个断点所在染色体、位置和链向、融合方向、支持信号的序列计数、正常序列的计数、质量值信息。
77.根据第二方面,在一实施例中,提供一种检测结构变异的装置,包括:
78.计算模块,用于计算待测样本测序文库的最大插入片段长度;
79.信号提取模块,用于从待测样本的测序数据中提取su信号、dp信号、sr信号;
80.信号处理模块,用于对sr信号、su信号、dp信号进行处理,获得结构变异信息。
81.根据第三方面,在一实施例中,提供一种装置,包括:
82.存储器,用于存储程序;
83.处理器,用于通过执行所述存储器存储的程序以实现第一方面的方法。
84.根据第四方面,在一实施例中,提供一种计算机可读存储介质,所述介质上存储有程序,所述程序能够被处理器执行以实现第一方面的方法。
85.在一实施例中,本发明可检测基因组的结构变异,支持肿瘤基因组结构变异的检测。
86.在一实施例中,本发明分为信号提取和信号处理两方面,包含sr、su、dp三类信号,基于信号确定结构变异发生的染色体、断点、方向等信息。
87.实施例1
88.图1为本实施例1的结构变异信息检测流程图,包括如下步骤:
89.1、计算最大插入片段长度
90.对符合正态分布的插入片段长度计算均值和方差,均值与三倍方差的和记为最大插入片段长度,该统计量是样本特异性的。
91.2、本实施例基于比对文件提取信号,比对文件中的每行代表每条序列(reads)的比对情况,包含比对文件的第二列信息flag、比对文件的第四列信息cigar等,信号提取后分别定义为sr、su和dp。
92.2.1本实施例提取的su信号,是指同时满足如下条件的reads对:flag不是次级比对(secondary,由于多比对,一条reads可能比对到染色体多个位置,主要比对结果为primary)或补充比对(supplementary,由于嵌合比对,一条reads的大部分比对正常为representative,其他部分比对到别处),两条reads中的一条flag为未比对(unmap)或其matereads为未必对(munmap),且两条reads中的非未比对序列不是重复比对(dup),将符合条件的flag为unmap的一条reads写出到临时bam文件并排序,其中,该reads的名字用其mate reads的信息命名,该信息包含比对上的参考基因组的名字、开始位置、终止位置、比对质量。
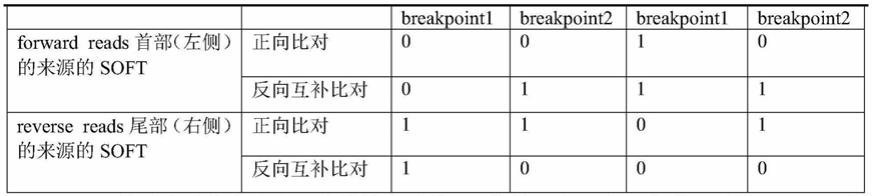
93.2.2本实施例提取的dp信号,是指同时满足如下条件的reads对:flag不是secondary、supplementary、dup、umap或munmap,两条reads比对上两个不同的染色体或两条reads对应的模板链长度大于最大插入片段长度,将符合条件的reads对写出到临时bam文件并排序,其中,reads的名字用其mate reads的信息命名,该信息包含比对上的参考基因组的名字、开始位置、终止位置、比对质量。
94.2.3本实施例提取的sr信号,是指不满足su和dp提取条件,且满足如下条件的reads对:cigar的首部或尾部是soft clip类型的reads,将符合条件的reads的soft clip信息写出到临时文本文件,其中,该soft clip信息包含soft clip位于首部或尾部、比对上的参考基因组的名字和比对位置、比对方向、soft clip序列、soft clip碱基质量、reads比对质量。
95.3、对soft clip信息的处理
96.3.1合并soft clip信息构建sr信号
97.对包含soft clip信息的临时文本文件按照soft clip位于首部或尾部、比对上的参考基因组的位置信息做初步排序,将soft clip位于首部或尾部、比对上的参考基因组的名字、比对位置、比对方向都相同的soft clip作为一簇,这种簇可以分为两种类型:对正向
序列(forward reads)首部的soft clip簇类型,按照soft clip逆序、去重计数、排序后构建新的soft clip集合;对反向序列(reverse reads)尾部的soft clip簇类型,按照soft clip原序、去重计数、排序后构建新的soft clip集合。
98.构建的有序的soft clip集合,符合如下条件可作为独立的sr信号:前后soft clip比较,若soft clip变短或没有前缀关系,则可将该soft clip及其计数(为1)作为独立的sr信号;若soft clip变长且符合前缀关系,则推举最长的soft clip及其计数(包含有前缀关系的softclip,个数大于1)作为独立的sr信号。将sr信号过滤掉小于20bp的部分后,写出到临时fastq文件,这里将临时fastq文件中的每条记录定义为soft以区别合并前的原始soft clip,其中,soft名字包含比对上的参考基因组的名字和比对位置、soft位于首部或尾部、比对方向、soft计数、比对质量、第几个soft。该过程也记录所有soft的计数和长度结果,其索引即为第几个soft,用于后续去重合并。
99.3.2对临时fastq文件的soft进行单端重比对
100.3.3从重比对结果中提取详细的结构变异信息
101.不考虑flag是secondary、supplementary、dup、umap或munmap的比对结果,从soft所在reads信息(记为breakpoint1,即第一断点)和soft重比对信息(记为breakpoint2,即第二断点)这两个信息内,就可以计算两个候选断点的初步信息:断点所在染色体、断点位置、断点soft所在reads和soft重比对的链向(is_reverse)、融合方向(is_gener),可以分为四种情况(如表1所示,0表示否,1表示是):forward reads首部(左侧)的来源的soft重比对后为正向比对到breakpoint2的左侧或反向互补比对到breakpoint2的右侧,reverse reads尾部(右侧)的来源的soft重比对后为正向比对到breakpoint2的右侧或反向互补比对到breakpoint2的左侧,进而还可以确定结构变异发生的细节,比如由断点所在染色体、断点位置、断点soft比对链向来确定结构变异的类型。
102.表1soft
[0103][0104][0105]
soft及其重比对信息确定了该结构变异的sr信号,在两个断点附近区域(soft所在侧延伸10bp、soft重比对侧延伸最大插入片段长度-2*测序reads长度,作为查找区间)寻找符合条件的dp信号作为补充:dp类型的两条reads对应的比对染色体和比对位置与breakpoint1和breakpoint2一致、链向一致(以soft所在reads的mate reads链向作为breakpoint1附近链向),确定dp信号的质量值和支持个数,找到的dp支持在后面dp信号处理时不再考虑,若未找到dp支持且soft的计数小于2,则过滤掉该sr信号。
[0106]
在breakpoint1附近区域(断点处向左flank最大插入片段长度到断点,作为查找区间)寻找符合条件的正常reads:不计算flag是secondary、supplementary、dup、umap或munmap的比对reads,和模板链长度大于最大插入片段长度或比对到不同染色体的reads,
基于比对起始位置在查找区间内的reads个数来确定正常reads的支持个数。
[0107]
3.4汇总
[0108]
由于多个soft确定的两个断点可能一样,需要合并为一个:以两个断点所在染色体、断点位置、断点soft所在reads和soft重比对的链向(is_reverse)、融合方向(is_gener)为索引进行合并,并更新sr信号支持个数。
[0109]
4、对su信号的处理
[0110]
将提取的su信号重比对后做局部组装,获得结构变异信息。由于组装后回比到参考基因组的序列很少,所以su信号处理后检出的结构变异也很少。
[0111]
5、对dp信号的处理
[0112]
结构变异附近的比对情况为:forward reads位于可能结构变异断点的左侧、reverse reads位于可能结构变异断点的右侧,基于此来区分forward和reverse链对dp信号的reads(已基于比对起始位置排过序)进行聚簇:相邻reads比较,若比对到相同染色体且比对起始位置相差小于等于200bp,则完成第一次聚簇,此时确定第一候选断点;对此次结果按照固定次序再次排序(这里比对位置和mate比对位置比较,按照整个基因组水平排序,可能对应mate为多簇,需排序),对mate部分聚簇,相邻序列比较,若比对到相同染色体且比对起始位置相差≤200bp,则完成第二次聚簇,此时确定第二候选断点;这样,两个断点附近配对成簇的dp信号得以初步确定。第二候选断点可能不止一个,若前两次聚簇后的序列个数≥5,则进行第三次聚簇,此时如果多个第二候选断点若相差》200bp,则分开成簇,再次返回序列个数≥5,获得最终的dp信号。
[0113]
若sr和su信号处理时已标记过,该dp也会跳过。
[0114]
确定正常reads的支持个数(同sr信号),以多的一侧作为breakpoint1的来源。
[0115]
dp信号的两个断点所在染色体、断点位置、链向、融合方向得以确定,进而确定结构变异的类型。
[0116]
6、结果写出
[0117]
将基于sr、su和dp信号确定的结构变异的信息,包含两个断点所在染色体、位置和链向、融合方向、支持信号的reads计数、正常reads的计数、质量值信息等,写出到最终结果文件。
[0118]
取panel样本1048例(重点关注其中1992个阳性sv集),提取dna建库进行芯片捕获测试,测序数据比对到人类参考基因组hg19上,然后采用本实施例的结果变异比对软件分析,检出率达到99.23%。
[0119]
本领域技术人员可以理解,上述实施方式中各种方法的全部或部分功能可以通过硬件的方式实现,也可以通过计算机程序的方式实现。当上述实施方式中全部或部分功能通过计算机程序的方式实现时,该程序可以存储于一计算机可读存储介质中,存储介质可以包括:只读存储器、随机存储器、磁盘、光盘、硬盘等,通过计算机执行该程序以实现上述功能。例如,将程序存储在设备的存储器中,当通过处理器执行存储器中程序,即可实现上述全部或部分功能。另外,当上述实施方式中全部或部分功能通过计算机程序的方式实现时,该程序也可以存储在服务器、另一计算机、磁盘、光盘、闪存盘或移动硬盘等存储介质中,通过下载或复制保存到本地设备的存储器中,或对本地设备的系统进行版本更新,当通过处理器执行存储器中的程序时,即可实现上述实施方式中全部或部分功能。
[0120]
以上应用了具体个例对本发明进行阐述,只是用于帮助理解本发明,并不用以限制本发明。对于本发明所属技术领域的技术人员,依据本发明的思想,还可以做出若干简单推演、变形或替换。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1