核酸测序数据的质量评估方法和筛选方法与流程
1.本发明涉及核酸测序数据的质量评估方法和筛选方法,属于基因测序领域。
背景技术:
2.高通量测序是一种同时对成千上万条dna序列进行序列测定的技术,被广泛应用于基础生物学和医学研究、以及体外诊断。通常,高通量测序仪除了输出所测dna序列之外,还会给输出的每一个碱基一个质量值,以表征该碱基的测量准确度。通常,这个质量值以phred的形式给出,即当该碱基的测量准确度为d时,所给质量值为:
3.q=-10 log
10
(1-p)
4.例如,质量值10对应准确度90%,质量值20对应准确度99%,质量值30对应准确度99.9%。
5.目前,高通量测序领域普遍使用的质量值评估方法为phred算法(ewing b,green p.base-calling of automated sequencer traces using phred.ii.error probabilities.genome res.8:186-194(1998))。phred算法被普遍应用于dna测序领域中,但不同测序技术所使用的预测器不同。例如,iontorrent使用的预测器有:碱基在序列中的位置;碱基所处同源多聚物的长度;碱基所处测序信号接近整数的程度;碱基所处测序信号的失相程度等。illumina也公开了一种使用神经网络的质量评估方法,即训练一个神经网络,通过预测器的值来预测碱基的质量,illumina的质量预测器值包括在线重叠、纯度、定相、start5、六聚体得分、基序累积、端值(endiness)、近似均聚物、强度衰减、倒数第二纯化度、与背景的信号重叠(sowb)和/或偏移的纯度g调整等(专利cn202080005431.0)。
6.一个好的打分算法,应当具有较高的区分度,即能够把高质量的碱基筛选出来,并给它打高分,把低质量的碱基筛选出来,并打低分。而利用现有算法对纠错码测序的测序数据进行质量评估时,存在区分度不足/较低的问题。因此,需要开发一种适用于纠错码测序数据且具有更高区分度的碱基质量评估方法。
7.高通量测序仪一次运行可产生大量的数据。然而,由于光学成像、信号采集、dna扩增产生的多克隆、化学反应、机械系统、算法处理等方面的缺陷及噪音的干扰,并非所有产生的数据均为高质量的数据。若下机后提供给用户的数据中包含低质量的、不可信的测序数据,则可能影响用户对样品的分析,造成假阳性或假阴性等误判,严重的可能影响临床诊断结果,延误病情。因此,对测序数据进行筛选、保留并输出高质量测序数据是高通量测序的必要步骤。
8.illumina采用“纯净度”来筛选其测序数据(美国专利us13006206)。具体地,illumina给四种碱基分别标记不同的荧光,测序时荧光最强的碱基即为所测碱基,其纯净度被定义为四种荧光中最强的荧光强度除以四种荧光强度之和,并认为纯净度高于给定阈值的序列为高质量序列。ion torrent利用两个指标来筛选其测序数据:ssq(校正信号与其取整值间的欧氏距离)和ppf(非零信号的比例)(专利ep19181402,wous11067959),认为这两个指标均小于给定阈值的序列为高质量序列。
9.在实际应用中,普遍利用高通量测序仪给所测dna序列的碱基打的质量值来筛选测序数据。例如fastp软件会对每条序列的质量值作移动平均,并在均值较低处对序列进行切除。也就是说筛选过程必须在得到碱基对应的质量值之后进行,增加了等待时间,有需要开发一种不依赖于碱基测序质量值的测序数据筛选方法。
技术实现要素:
10.一方面,本发明提供一种核酸测序数据的质量评估方法,其特征在于,包括:
11.a)对参考核酸样品进行测序,获得一组测序信号s;用相同的测序方法对待测核酸样品进行测序,获得一组测序信号s’;
12.b)对所述测序信号s进行信号校正,获得校正信号c,所述校正信号c可直接转换成碱基序列;
13.c)将转换出的碱基序列比对到参考序列上,得到比对结果,再根据所述比对结果将碱基标记为测序正确或测序错误;
14.d)对所述测序正确或测序错误的碱基,比较其在测序信号s和校正序列c中对应部分的信号差异,并建立信号差异和碱基测序准确率之间的联系;
15.e)用与步骤b)相同的校正方式对所述测序信号s’进行信号校正,获得校正信号c’,再将c’转换成碱基序列;
16.f)对转换出的碱基序列中的每个碱基,比较其在s’和c’中对应部分的信号差异,利用步骤d)中建立的信号差异和碱基准确率之间的联系,预测该碱基的测序准确率。
17.根据优选的实施方式,所述测序产生的信号类型可以是光信号,也可以是电信号;所述光信号可以是单色的,也可以是多色的。
18.根据优选的实施方式,所述待测核酸包括dna,或者rna或其他类型的核酸分子。
19.根据优选的实施方式,所述参考核酸样品所属物种的基因组序列已知;所述参考核酸是dna时,所述参考序列为参考核酸所属物种的基因组序列;所述参考核酸是rna时,所述参考序列为参考核酸所属物种的转录组序列。
20.根据优选的实施方式,将转换出的碱基序列比对到参考序列上,得到比对结果,从中进一步筛选出高质量比对的碱基序列,再将所述高质量比对的碱基序列中的碱基标记为测序正确或测序错误;所述的高质量比对,需要根据所用的比对软件或算法来具体选择质量值范围。
21.根据优选的实施方式,所述测序信号是测序仪直接采集到的信号,或者是经过归一化后的信号;相对应地,对测序信号的校正过程,可以是失相校正,所述校正信号是失相校正后的信号。
22.根据优选的实施方式,在纠错码测序或采用了精准化学读出的solid等测序技术中,所述测序信号可以是失相校正后的信号;相对应地,对测序信号的校正过程,可以是纠错校正过程,所述校正信号是纠错校正后的信号。
23.根据优选的实施方式,在采用环形一致性测序模式的单分子实时测序中,或利用dna复制、反复多次测序来提高准确度的纳米孔测序技术中,所述测序信号可以是原始序列;相对应地,对测序信号的校正过程,可以是求一致性序列的过程。
24.根据优选的实施方式,所述建立信号差异和碱基准确率之间联系的方法,是构建
信号差异和碱基准确率之间的对照表。
25.根据优选的实施方式,所述建立信号差异和碱基准确率之间联系的方法,是将一个或多个预测器划分成若干个区间,统计每个区间内碱基的准确率及准确率对应的质量值;评估的方法可以是计算所测核酸中的每个碱基落入哪个预测器的区间,再将该区间对应的质量值赋给该碱基。
26.根据优选的实施方式,所述建立信号差异和碱基准确率之间联系的方法及对应的评估方法,是phred算法。
27.根据优选的实施方式,所述建立信号差异和碱基准确率之间联系的方法,是机器学习。
28.根据优选的实施方式,可以比较测序信号s和校正信号c的多种不同差异,作为多个预测器来共同评估碱基质量;设s=(s1,s2,...,sn),c=(c1,c2,...,cn),则所述测序信号s和校正信号c的多种不同差异包括:
29.1)
30.2)
31.3)
32.4)max|s
i-ci|
33.5)min|s
i-ci|
34.6)
35.7)
36.根据优选的实施方式,所述比较测序信号s和校正信号c的差异,所述差异可以是两种信号中一部分的差异,包括:
37.1)全部子信号的差异;
38.2)前若干个子信号的差异;
39.3)后若干个子信号的差异;
40.4)中间若干个子信号的差异;
41.5)奇数编号的子信号的差异;
42.6)偶数编号的子信号的差异;
43.7)s或c中大于给定阈值的子信号的差异;
44.8)s或c中小于给定阈值的子信号的差异;
45.9)以上选项的组合,包括但不限于前若干个奇数编号的子信号的差异。
46.根据优选的实施方式,可以比较测序信号s和校正信号c的局部差异;所述局部差异是指对于编号为i的测序信号si和校正信号ci,其前后若干个子信号(s
i-m
,s
i-m+1
,s
i-m+2
,
…
,s
i+m-1
,s
i+m
)和(c
i-m
,c
i-m+1
,c
i-m+2
,
…
,c
i+m-1
,c
i+m
)之间的差异,即对每个子信号均可计算一个局部差异,得到一组局部差异;m为小于i的整数。
47.根据优选的实施方式,在使用测序信号s和校正信号c的差异的基础上,利用其他预测器来共同评估碱基质量,所述其他预测器包括但不限于:
48.1)碱基在序列中的位置;
49.2)碱基所处同源多聚物的长度;
50.3)碱基在所处同源多聚物中的位置;
51.4)碱基所处测序信号接近整数的程度;
52.5)碱基所处测序信号的失相程度;
53.6)碱基所处测序信号的衰减程度;
54.7)碱基所处测序信号在校正过程中估计所得的参数,包括单位信号、背景信号、衰减系数、超前系数、滞后系数。
55.根据优选的实施方式,在纠错码测序中,在使用测序信号s和校正信号c的差异的基础上,结合碱基所处简并多聚物的长度作为预测器,来共同评估碱基质量。
56.根据优选的实施方式,在纠错码测序中,在使用测序信号s和校正信号c的差异的基础上,结合碱基所处简并多聚物中较多的那一种碱基的数目作为预测器,来共同评估碱基质量。
57.本发明提供一种核酸测序数据的质量评估系统,其特征在于,包括:
58.处理器,存储器,以及用于对核酸测序数据进行质量评估的程序,所述程序包括如下指令:
59.a)将参考核酸样品测序获得的测序信号s进行信号校正,获得校正信号c,所述校正信号c可直接转换成碱基序列;
60.b)将转换出的碱基序列比对到参考序列上,得到比对结果,再根据所述比对结果将碱基标记为测序正确或测序错误;
61.c)对所述测序正确或测序错误的碱基,比较其在测序信号s和校正信号c中对应部分的信号差异,并建立信号差异和碱基准确率之间的联系;
62.d)将待测核酸样品测序获得的新的一组测序信号s’用与步骤a)相同的校正方式进行信号校正,获得校正信号c’,再将c’转换为碱基序列;
63.e)对转换出的碱基序列中的每个碱基,比较其在s’和c’中对应部分的信号差异,利用步骤c)中建立的信号差异和碱基准确率之间的联系,预测该碱基的测序准确率。
64.另一方面,本发明提供一种核酸测序数据的筛选方法,其特征在于,包括:
65.a)对待测核酸样品进行测序,获得一组测序信号;
66.b)对所述测序信号进行信号校正,获得校正信号,所述校正信号可直接转换为碱基序列;
67.c)对转换出的碱基序列,比较其在测序信号和校正信号中对应部分的信号差异;
68.d)若测序信号和校正信号之间的所述信号差异大于给定的阈值,则丢弃该测序信号,否则予以保留。
69.根据优选的实施方式,所述测序产生的信号类型可以是光信号,也可以是电信号;所述光信号可以是单色的,也可以是多色的。
70.根据优选的实施方式,所述待测核酸包括dna,或者rna或其他类型的核酸分子。
71.根据优选的实施方式,所述测序信号是测序仪直接采集到的信号,或者是经过归一化后的信号;相对应地,对测序信号的校正过程,可以是失相校正,所述校正信号是失相校正后的信号。
72.根据优选的实施方式,在纠错码测序或采用了精准化学读出的solid等测序技术中,所述测序信号可以是失相校正后的信号;相对应地,对测序信号的校正过程,可以是纠错校正过程,所述校正信号是纠错校正后的信号。
73.根据优选的实施方式,在采用环形一致性测序模式的单分子实时测序中,或利用dna复制、反复多次测序来提高准确度的纳米孔测序技术中,所述测序信号可以是原始序列;相对应地,对测序信号的校正过程,可以是求一致性序列的过程。
74.根据优选的实施方式,可以比较测序信号和校正信号的多种不同差异;设测序信号=(s1,s2,...,sn),校正信号=(c1,c2,...,cn),则所述测序信号和校正信号的多种不同差异包括:
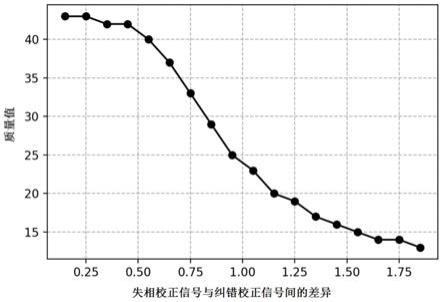
75.1)
76.2)
77.3)
78.4)max|s
i-ci|
79.5)min|s
i-ci|
80.6)
81.7)
82.根据优选的实施方式,所述比较测序信号和校正信号的差异,所述差异可以是两种信号中一部分的差异,包括:
83.1)全部子信号的差异;
84.2)前若干个子信号的差异;
85.3)后若干个子信号的差异;
86.4)中间若干个子信号的差异;
87.5)奇数编号的子信号的差异;
88.6)偶数编号的子信号的差异;
89.7)s或c中大于给定阈值的子信号的差异;
90.8)s或c中小于给定阈值的子信号的差异;
91.9)以上选项的组合,包括但不限于前若干个奇数编号的子信号的差异。
92.根据优选的实施方式,可以比较测序信号和校正信号的局部差异;所述局部差异是指对于编号为i的测序信号si和校正信号ci,其前后若干个子信号(s
i-m
,s
i-m+1
,s
i-m+2
,
…
,s
i+m-1
,s
i+m
)和(c
i-m
,c
i-m+1
,c
i-m+2
,...,c
i+m-1
,c
i+m
)之间的差异,即对每个子信号均可计算一个局部差异,得到一组局部差异;m为小于i的整数;如果所述一组局部差异中,某个局部差异大于给定的阈值,则在序列中切除局部差异过大的部分,输出截短后的序列;如果截短后的序列的长度小于预设值,可以丢弃该整条序列。
93.根据优选的实施方式,可以比较测序信号和校正信号的多种不同差异,综合判断后决定丢弃还是保留待筛选序列。
94.本发明还提供一种核酸测序数据的筛选系统,其特征在于,包括:
95.处理器,存储器,以及用于对核酸测序数据进行筛选的程序,所述程序包括如下指令:
96.a)将待测核酸样品测序获得的测序信号进行信号校正,获得校正信号,所述校正信号可直接转换为碱基序列;
97.b)对转换出的碱基序列,比较其在测序信号和校正信号中对应部分的信号差异;
98.c)若测序信号和校正信号之间的所述信号差异大于给定的阈值,则丢弃该测序信号,否则予以保留。
99.本发明的有益效果
100.本发明公开的测序数据的质量评估方法和筛选方法,将测序信号和校正信号之间的信号差异作为重要的判断标准,具有如下优势:
101.1.本发明选取测序信号和校正信号的差异作为预测器,该预测器含有表征碱基准确性最大的信息量,适用于纠错码测序。
102.2.与现有方法相比,本发明的方法具有较高区分度,所评估出的质量值最高可达45,超过illumina的41、roche 454的40。
103.3.本发明的测序数据筛选方法,不需要先得到每个碱基对应的质量值,只需要利用测序信号和校正信号之间的差异,来筛选测序数据,节省了测序时间。
104.4.本发明的测序数据筛选方法可以利用失相校正后的信号和纠错校正后的信号之间的差异,来评估或筛选测序数据,这是首个专门适用于纠错码测序的测序数据评估和筛选方法。本方法适用范围广泛,适用于包括纠错码测序在内的多种测序方法得到的测序数据。
附图说明
105.为了更清楚地说明本发明实施例,下面将对实施例中所需要使用的附图进行简单的介绍。显而易见地,下面描述中的附图仅是符合本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他实施例对应的附图。
106.图1.失相校正信号与纠错校正信号间的差异与碱基质量值的对应关系图。
107.图2.利用质量评估表对待测核酸进行纠错码测序数据的评估结果区分度图。
108.图3.对本发明评估方法打分的结果准确性的评估图。
109.图4.测序信号与校正信号的信号差异密度分布图。
具体实施方式
110.在高通量测序技术中,为了放大测序信号,在测序前普遍将待测dna分子进行扩增,形成dna团簇。理想情况下,同一dna分子扩增形成的团簇中的所有分子在测序的每一轮都应被合成出同样长度的新生链,这样测序信号与被合成的新生链一一对应,可以获得序列信息。然而,实际测序中,一个不可避免的限制因素是失相(dephasing),即一批相同序列的dna分子的新生链在合成过程中失去同步性。这主要是由预期之外的核苷酸被聚合酶掺入或不完全延伸所造成。失相包括两种类型:由于错配或杂质核苷酸的存在,dna团簇中一部分分子的延伸长度高于预期值,造成“超前”(lead);一部分分子因反应不完全而未被聚
consensus sequencing,inc-seq)为代表的技术,该方法首先需要环化模板dna分子,然后利用滚环扩增来扩增分子,生成由多个重复单元组成的环状分子,这些环状分子随后在测序前被打断成线性dna链。对来自同一原始序列的dna reads进行多次测序,可以通过计算合并这些信息,得到原始dna序列的准确read。此外,利用dna复制、反复多次测序来提高准确度的纳米孔测序技术还包括circ-seq、r2c2等技术。
124.比对
125.比对(align或alignment):“比对”是生物信息学中的常见概念,在生物信息学中,比对经常用于比较不同核酸之间或者不同蛋白质之间的相似性。本发明中的比对指的是将测序得到的碱基序列和参考序列进行比较,从而确定测序所得的碱基序列正确与否。常用的序列比对算法及软件包括但不限于,例如的,smith-waterman算法、bowtie、bwa、soap、needleman-wunch算法、bowtie2、blast、eland、tmap、maq、minimap2、shrimp等。
126.比对质量
127.利用前述常用的比对软件或算法将测序得到的碱基序列比对到参考序列上,以bowtie2为例,bowtie2使用打分机制,而不是完全的mismatch对比对质量进行衡量,分值越高说明正确比对的概率越大。本发明中所述的高质量比对,需要根据所用的比对软件或算法来具体选择质量值范围;例如的,当使用bwa进行序列比对时,高质量比对的碱基序列指的是,比对质量大于0、或大于等于10、或大于等于20、或大于等于30、或大于等于40、或大于等于50、或大于等于60的碱基序列。
128.归一化信号
129.即经过归一化后的信号,原始测序信号经过衰减校正后得到较为准确的单位信号,每个测序位点衰减校正后的测序信号与此位点的单位信号的比值即为此位点在每个测序轮中的归一化信号。
130.预测器
131.预测器,是碱基的特征,本发明中的预测器包括,例如:测序信号s和校正信号c之间的差异,碱基在序列中的位置,碱基所处同源多聚物的长度,碱基在所处同源多聚物中的位置,碱基所处简并多聚物中较多的那一种碱基的数目,碱基所处测序信号接近整数的程度,碱基所处测序信号的失相程度,碱基所处测序信号的衰减程度,碱基所处测序信号在校正过程中估计所得的参数,如单位信号、背景信号、衰减系数、超前系数、滞后系数,等。根据上述与碱基读出相关联的特征(预测器)为碱基识别分配错误概率。在这种方法中,首先用到一个训练数据集,将此训练数据集中的特征与一个已知的错误率匹配(错误率通常由与参考基因组的比对决定)。然后,对于一个目标数据集,可将上述方法应用于将此特征映射到错误概率(或质量值,其是错误概率的对数变换)。也就是说预测器并不能帮助提高碱基读出的质量,而是可以帮助区分低质量和高质量的碱基读出。现有技术中,利用多种不同的方法,例如位置特异性错误概率、phred、pyrobayes、逻辑回归、svm、矢量量化等,实现了将错误概率分配给相应的碱基读出的结果。
132.简并多聚物的长度
133.简并测序是一种多碱基测序,区别于单碱基测序每轮反应只延伸一个核苷酸分子,多碱基测序每轮反应延伸的核苷酸可能是多个,测序反应释放的荧光信号强度与释放的荧光基团数目成正相关,在没有衰减和失相的理想条件下,每轮反应的荧光信号反映了
该轮延伸的碱基数,被称为简并多聚物的长度(degenerate polymer length,dpl)。
134.简并碱基
135.本发明中,按照iupac符号命名规则(nucleic acid notation),使用下面表1的字母表示简并碱基,例如字母m表示a和/或c。
136.表1
137.字母所代表的碱基ma/ckg/tra/gyc/twa/tsc/gbc/g/tda/g/tha/c/tva/c/g
138.碱基质量值
139.该数值可以是碱基识别出错概率的整数映射。目前通用的表示形式为:公式q=-10 log
10
(1-p),式中:p为碱基的测量准确度,其值越高表明碱基识别越可靠,碱基测错的可能性越小。例如,质量值为20(常写作q20),计算得p=0.99,测序正确率99%,质量值为30(常写作q30),计算得p=0.999,测序正确率99.9%。目前,illumina测序仪所能得到的最高质量值为41、roche454得到的最高质量值为40。可以理解的,该质量值只是碱基测序准确率的表示形式,表示形式本身并不重要,也可以直接用准确率来表示。
140.2.发明详述
141.具体的,本发明的第一方面提供一种核酸测序数据的质量评估方法,其特征在于,包括:
142.a)对参考核酸样品进行测序,获得一组测序信号s;用相同的测序方法对待测核酸样品进行测序,获得一组测序信号s’;
143.b)对所述测序信号s进行信号校正,获得校正信号c,所述校正信号c可直接转换成碱基序列;
144.c)将转换出的碱基序列比对到参考序列上,得到比对结果,再根据所述比对结果将碱基标记为测序正确或测序错误;
145.d)对所述测序正确或测序错误的碱基,比较其在测序信号s和校正序列c中对应部分的信号差异,并建立信号差异和碱基测序准确率之间的联系;
146.e)用与步骤b)相同的校正方式对所述测序信号s’进行信号校正,获得校正信号c’,再将c’转换成碱基序列;
147.f)对转换出的碱基序列中的每个碱基,比较其在s’和c’中对应部分的信号差异,利用步骤d)中建立的信号差异和碱基准确率之间的联系,预测该碱基的测序准确率。
148.根据优选的实施方式,所述测序方法包括但不限于:双脱氧核苷酸终止法
(sanger),焦磷酸测序法(454),半导体测序法(ion torrent),可逆循环终止法(illumina),荧光发生测序法(赛纳生物科技),纠错码测序法(ecc),模糊测序法(专利cn201611088606.0),联合探针锚定连接法(华大),寡核苷酸连接检测测序法(solid),单分子荧光测序法(helicos),单分子实时测序法(pacbio),纳米孔测序法(nanopore)等;还包括基于各种测序方法的升级技术,例如pacbio公司在单分子实时测序法基础上改进的环形一致性测序法(circular consensus sequencing,ccs),即通过对同一个分子反复测序,可以计算出一致性序列,进而提高准确性;基于纳米孔测序法的分子内连接的纳米孔一致性测序(intramolecular-ligated nanopore consensus sequencing,inc-seq)等。
149.根据优选的实施方式,所述待测核酸包括dna,或者rna或其他类型的核酸分子,例如,x核酸(xenonucleic acids)(或称xnas)、肽核酸(pna)、锁式核糖核酸(lna)等。待测核酸分子的类型不改变本发明的核心内容。
150.在一些实施方式中,对参考核酸样品和待测核酸样品同时进行测序,在可选的实施方式中,对参考核酸样品和待测核酸样品可以不同时测序,例如的,可以先对参考核酸样品进行测序,再用相同的测序方法对待测核酸样品进行测序;测序的顺序并不重要,必须要满足的条件是上述核酸样品使用同种测序方法进行测序,例如,都使用纠错码测序。
151.根据优选的实施方式,所述测序产生的信号类型可以是光信号,也可以是电信号;所述光信号可以是单色的,也可以是多色的。例如,对于半导体测序法,测序产生的信号是电信号;对于焦磷酸测序法、可逆循环终止法、荧光发生测序法等,产生的测序信号是荧光信号,即光信号。
152.在具体的实施方式中,将校正信号转换得到的碱基序列比对到参考序列上,得到比对结果,再根据所述比对结果将碱基标记为测序正确或测序错误;优选的,从比对结果中进一步筛选出高质量比对的碱基序列,再将所述高质量比对的碱基序列中的碱基标记为测序正确或测序错误,忽略无法确定的碱基(即无法成功比对到参考序列上的碱基或比对质量较低的碱基)。根据比对结果,将比对结果为“匹配”的碱基标记为“测序正确”,将比对结果为“错配”、“插入”或“缺失”的碱基标记为“测序错误”。本发明中所述的高质量比对,需要根据所用的比对软件或算法来具体选择质量值范围;例如的,当使用bwa进行序列比对时,高质量比对的碱基序列指的是,比对质量大于0、或大于等于10、或大于等于20、或大于等于30、或大于等于40、或大于等于50、或大于等于60的碱基序列。
153.本发明中,当参考核酸是dna时,参考序列为参考核酸所属物种的基因组序列;当参考核酸是rna时,参考序列为参考核酸所属物种的转录组序列。在参考序列的选择上需要注意,参考核酸样品必须是所属物种的基因组序列已知的,且应选择碱基突变率低的,便于确定测序序列与参考序列之间的不同到底是由于测序错误还是正常的碱基突变造成的。例如,标准核酸样品可以选自λ噬菌体,大肠杆菌,酿酒酵母等的核酸。
154.根据优选的实施方式,测序信号可以是测序仪直接采集到的信号,也可以是经过归一化后的信号;相对应地,对测序信号的校正过程,可以是失相校正,校正信号是失相校正后的信号;对测序信号s进行的信号校正与对测序信号s’进行的校正相同。
155.根据优选的实施方式,在采用环形一致性测序(circular consensus sequencing)的单分子实时测序中,或某些利用dna复制、反复多次测序来提高准确度的纳米孔测序(如circ-seq、inc-seq、r2c2等)中,测序信号可以是原始序列;相对应地,对测序
信号的校正过程,可以是求一致性序列的过程;对测序信号s进行的信号校正与对测序信号s’进行的校正相同。
156.根据优选的实施方式,在纠错码测序、采用了精准化学读出(exact call chemistry)技术的solid等测序技术中,测序信号可以是失相校正后的信号;相对应地,对测序信号的校正过程,可以是纠错校正过程。对测序信号s进行的信号校正与对测序信号s’进行的校正相同。对于纠错码测序,具体的纠错校正过程可以参见专利cn201510944878.5,纠错校正过程本身并不是本发明的重点,由纠错校正后的校正信号与校正前的失相校正信号之间的信号差异,才是本发明重点关注的内容。所述信号差异的表征方式可以分为两大类,第一类是一般差异,一般差异可以是全部子信号或部分子信号的差异,第二类是局部差异,具体描述见下文。需要了解的是,信号差异的表征方式仅仅是数学公式的具体运用,并不影响本发明的核心内容。
157.根据优选的实施方式,可以比较测序信号s和校正信号c的多种不同差异,作为多个预测器来共同评估碱基质量;设s=(s1,s2,...,sn),c=(c1,c2,...,cn),则所述测序信号s和校正信号c的多种不同差异包括:
158.1)
159.2)
160.3)
161.4)max|s
i-ci|
162.5)min|s
i-ci|
163.6)
164.7)
165.也可以是取测序信号s和校正信号c的一部分子信号利用上述公式来计算信号差异,所述子信号包括以下多种类型:
166.1)全部子信号的差异;
167.2)前若干个子信号的差异;
168.3)后若干个子信号的差异;
169.4)中间若干个子信号的差异;
170.5)奇数编号的子信号的差异;
171.6)偶数编号的子信号的差异;
172.7)s或c中大于给定阈值的子信号的差异;
173.8)s或c中小于给定阈值的子信号的差异;
174.9)以上选项的组合,包括但不限于前若干个奇数编号的子信号的差异。
175.在优选的实施方式中,可以比较测序信号s和校正信号c的局部差异;所述局部差异指对于编号为i的测序信号si和校正信号ci,其前后若干个子信号(s
i-m
,s
i-m+1
,s
i-m+2
,
…
,s
i+m-1
,s
i+m
)和(c
i-m
,c
i-m+1
,c
i-m+2
,
…
,c
i+m-1
,c
i+m
)之间的差异,即对每个子信号均可计算一个局部差异,得到一组局部差异;m为小于i的整数。
176.在某些实现中,可以比较测序信号s和校正信号c的多种不同差异,作为多个预测器来共同评估碱基质量。
177.在某些实现中,在使用测序信号s和校正信号c的差异的基础上,可以利用其他已报道的预测器来共同评估碱基质量,这些预测器包括但不限于:
178.1)碱基在序列中的位置;
179.2)碱基所处同源多聚物的长度;
180.3)碱基在所处同源多聚物中的位置;
181.4)碱基所处测序信号接近整数的程度;
182.5)碱基所处测序信号的失相程度;
183.6)碱基所处测序信号的衰减程度;
184.7)碱基所处测序信号在校正过程中估计所得的参数,如单位信号、背景信号、衰减系数、超前系数、滞后系数。
185.在优选的实施方式中,建立信号差异和碱基准确率之间联系的方法,可以是构建信号差异和碱基准确率之间的对照表。例如,当信号差异为0.05时,对应的准确率为99.9%;当信号差异为0.1时,对应的准确率为99%;以此类推,构建出一张对照表。基本步骤的第f)步中,只需查询该对照表中信号差异所对应的准确率,即可预测被测碱基的准确率。在某些实现中,该对照表中的信号差异可以以区间的形式表示,例如,当信号差异处于0.05到0.08之间时,对应的准确率均为99.9%。
186.基本步骤中,建立信号差异和碱基准确率之间联系的方法,可以是将一个或多个预测器划分成若干个区间,统计每个区间内碱基的准确率及准确率对应的质量值;评估的方法可以是计算所测核酸中的每个碱基落入哪个预测器的区间,然后将该区间对应的质量值赋给该碱基。具体的,可以逐行查询质量评估表,若待评估碱基的预测器值均小于表中某行记录的预测器阈值,则将该行记录的质量值赋给该碱基,否则继续查询下一行。
187.基本步骤中,建立信号差异和碱基准确率之间联系的方法,及对应的评估方法,可以是phred算法(ewing b,green p.base-calling of automated sequencer traces using phred.ii.error probabilities.genome res.8:186-194(1998))。该方法分为训练和评估两步,其中训练步骤简要描述如下:
188.1.利用某种监督性方法,把每个碱基标记为正确或错误。
189.2.利用测序信号,对每个碱基都计算若干个预测器的值。设一共有m个预测器(p1,p2,...,pm),每个预测器的值均随错误率的上升而单调上升。
190.3.给每个预测器划分若干个阈值。设每个预测器分别划分了n1,n2,...,nm个阈值,则所有预测器一共被划分成了n=n1×
n2×
...
×
nm个区间。
191.4.计算每个区间所对应碱基的错误率。设某个区间的阈值为(t1,t2,...,tm),则该区间对应的碱基为预测器值满足p1≤t1,p2≤t2,...,pm≤tm的碱基。
192.5.挑选出错误率最低的一个区间,将该区间的预测器阈值、对应的错误率和质量值记入质量评估表中。
193.6.删除上一步中被挑选的区间及其对应的碱基。
194.7.若所有区间或所有碱基均已被删除完,则结束训练并输出质量评估表,否则回到第4步。
195.评估步骤简要描述如下:
196.1.对待评估的碱基,计算其预测器的值。
197.2.逐行查询质量评估表,若待评估碱基的预测器值均小于表中某行记录的预测器阈值,则将该行记录的质量值赋给该碱基,否则继续查询下一行。
198.3.若质量评估表查完了也未给待评估的碱基赋予质量值,则可以赋予一个统一的质量值,如0、10或20等。
199.基本步骤中,建立信号差异和碱基准确率之间联系的方法,可以是机器学习。具体地,以一个或多个预测器的值作为输入,碱基是否正确作为输出,利用机器学习的方法训练得到输入和输出间的一个分类器。评估的方法可以是计算待测dna中的碱基的预测器值,输入到所训练的分类器中,根据分类器的输出来得到碱基的准确率及其对应的质量值。根据分类器的输出来计算分类准确率是机器学习中的常用技术,常见的方式例如soft-max,即若分类器的输出中,表示“正确”和“错误”的输出分别为a和b,则准确率为:
[0200][0201]
在一些优选的实施方式中,对于纠错码测序,在使用测序信号s和校正信号c的差异的基础上,可以结合碱基所处简并多聚物的长度作为预测器,来共同评估碱基质量。例如,在序列acttgaaatc中,第5个碱基g处于三种简并多聚物ttg、gaaa、g中,对应的简并多聚物的长度分别是3、4、1。
[0202]
在一些优选的实施方式中,对于纠错码测序,在使用测序信号s和校正信号c的差异的基础上,可以结合碱基所处简并多聚物中较多的那一种碱基的数目作为预测器,来共同评估碱基质量。例如,在序列acttgaaatc中,第5个碱基g处于三种简并多聚物ttg、gaaa、g中,其中较多的那一种碱基分别为t、a、g,其数目分别为2、3、1。
[0203]
本发明还提供一种核酸测序数据的质量评估系统,其特征在于,包括:处理器,存储器,以及用于对核酸测序数据进行质量评估的程序,所述程序包括如下指令:
[0204]
a)将参考核酸样品测序获得的测序信号s进行信号校正,获得校正信号c,所述校正信号c可直接转换成碱基序列;
[0205]
b)将转换出的碱基序列比对到参考序列上,得到比对结果,再根据所述比对结果将碱基标记为测序正确或测序错误;
[0206]
c)对所述测序正确或测序错误的碱基,比较其在测序信号s和校正信号c中对应部分的信号差异,并建立信号差异和碱基准确率之间的联系;
[0207]
d)将待测核酸样品测序获得的新的一组测序信号s’用与步骤a)相同的校正方式进行信号校正,获得校正信号c’,再将c’转换为碱基序列;
[0208]
e)对转换出的碱基序列中的每个碱基,比较其在s’和c’中对应部分的信号差异,利用步骤c)中建立的信号差异和碱基准确率之间的联系,预测该碱基的测序准确率。
[0209]
在本发明的第一方面具体实施部分中所讨论的特征中的每个特征同样适用于本发明的核酸测序数据的质量评估系统的具体实施。如上所示,所有其他特征在此处不再重复,并且应被视为以引用方式重复。本领域普通技术人员将理解在这些具体实施中识别的特征可如何容易地与在其他具体实施中识别的基本特征组组合。
[0210]
按照领域内的一般做法,在碱基质量评估完成后,可进一步根据碱基质量值对所
测核酸序列进行筛选。例如,若一条序列上碱基的平均质量值低于某个给定的阈值,可以丢弃该序列,不予输出;若一条序列的尾部的碱基的平均质量值低于某个给定的阈值,可将尾部截去,仅输出头部高质量的部分。有很多工具可完成该步骤,例如fastp软件、fastqc软件等。但是这种筛选方法必须要在获得碱基质量值之后才能进行,势必要浪费大量的等待时间,对于有测序需求的用户来说,能够在尽可能短的时间内得到筛选后的高质量测序结果是其最期望的。因此,有必要开发一种无需对碱基质量进行打分即可进行高质量测序数据筛选的方法。
[0211]
本发明的另一方面提供一种核酸测序数据的筛选方法,其特征在于,包括:
[0212]
a)对待测核酸样品进行测序,获得一组测序信号;
[0213]
b)对所述测序信号进行信号校正,获得校正信号,所述校正信号可直接转换为碱基序列;
[0214]
c)对转换出的碱基序列,比较其在测序信号和校正信号中对应部分的信号差异;
[0215]
d)若测序信号和校正信号之间的所述信号差异大于给定的阈值,则丢弃该测序信号,否则予以保留。
[0216]
根据优选的实施方式,对待测核酸样品进行测序的方法包括但不限于:双脱氧核苷酸终止法(sanger),焦磷酸测序法(454),半导体测序法(ion torrent),可逆循环终止法(illumina),荧光发生测序法(赛纳生物科技),纠错码测序法(ecc),模糊测序法(专利cn201611088606.0),联合探针锚定连接法(华大),寡核苷酸连接检测测序法(solid),单分子荧光测序法(helicos),单分子实时测序法(pacbio),纳米孔测序法(nanopore)等。还包括基于各种测序方法的升级技术,例如pacbio公司在单分子实时测序法基础上改进的环形一致性测序法(circular consensus sequencing,ccs),即通过对同一个分子反复测序,可以计算出一致性序列,进而提高准确性;基于纳米孔测序法的分子内连接的纳米孔一致性测序(intramolecular-ligated nanopore consensus sequencing,inc-seq)等。
[0217]
根据优选的实施方式,所述待测核酸包括dna,或者rna或其他类型的核酸分子,例如,x核酸(xenonucleic acids)(或称xnas)、肽核酸(pna)、锁式核糖核酸(lna)等。待测核酸分子的类型不改变本发明的核心内容。
[0218]
根据优选的实施方式,所述测序产生的信号类型可以是光信号,也可以是电信号;所述光信号可以是单色的,也可以是多色的。例如,对于半导体测序法,测序产生的信号是电信号;对于焦磷酸测序法、可逆循环终止法、荧光发生测序法等,产生的测序信号是荧光信号,即光信号。
[0219]
根据优选的实施方式,测序信号可以是测序仪直接采集到的信号,也可以是经过归一化后的信号;相对应地,对测序信号的校正过程,可以是失相校正,所述校正信号是失相校正后的信号。
[0220]
根据优选的实施方式,在采用环形一致性测序(circular consensus sequencing)的单分子实时测序中,或某些利用dna复制、反复多次测序来提高准确度的纳米孔测序(如circ-seq、inc-seq、r2c2等)中,测序信号可以是原始序列;相对应地,对测序信号的校正过程,可以是求一致性序列的过程。
[0221]
根据优选的实施方式,在纠错码测序、采用了精准化学读出(exact call chemistry)技术的solid等测序技术中,测序信号可以是失相校正后的信号;相对应地,对
测序信号的校正过程,可以是纠错校正过程。对于纠错码测序,具体的纠错校正过程可以参见专利cn201510944878.5,纠错校正过程本身并不是本发明的重点,由纠错校正后的校正信号与校正前的失相校正信号之间的信号差异,才是本发明重点关注的内容。所述信号差异的表征方式可以分为两大类,第一类是一般差异,一般差异可以是全部子信号或部分子信号的差异,第二类是局部差异,具体描述见下文。需要了解的是,信号差异的表征方式仅仅是数学公式的具体运用,并不影响本发明的核心内容。
[0222]
根据优选的实施方式,可以比较测序信号和校正信号的多种不同差异,设测序信号=(s1,s2,...,sn),校正信号=(c1,c2,...,cn),则所述测序信号和校正信号的多种不同差异包括:
[0223]
1)
[0224]
2)
[0225]
3)
[0226]
4)max|s
i-ci|
[0227]
5)min|s
i-ci|
[0228]
6)
[0229]
7)
[0230]
以上是取测序信号和校正信号的全部子信号来计算差异,容易仿照上述公式给出如何取一部分子信号来计算差异。
[0231]
根据优选的实施方式,所述比较测序信号和校正信号的差异,这一差异可以是两种信号中一部分的差异,包括但不限于:
[0232]
1)全部子信号的差异;
[0233]
2)前若干个子信号的差异;
[0234]
3)后若干个子信号的差异;
[0235]
4)中间若干个子信号的差异;
[0236]
5)奇数编号的子信号的差异;
[0237]
6)偶数编号的子信号的差异;
[0238]
7)测序信号或校正信号中大于给定阈值的子信号的差异;
[0239]
8)测序信号或校正信号中小于给定阈值的子信号的差异;
[0240]
9)以上选项的组合,包括但不限于前若干个奇数编号的子信号的差异。
[0241]
根据优选的实施方式,可以比较测序信号和校正信号的局部差异;所述局部差异指对于编号为i的测序信号si和校正信号ci,其前后若干个子信号(s
i-m
,s
i-m+1
,s
i-m+2
,
…
,s
i+m-1
,s
i+m
)和(c
i-m
,c
i-m+1
,c
i-m+2
,
…
,c
i+m-1
,c
i+m
)之间的差异,即对每个子信号均可计算一个局部差异,得到一组局部差异;m为小于i的整数;若所述一组局部差异中,某个局部差异大于给定的阈值,则在序列中切除局部差异过大的部分,输出截短后的序列;所述阈值并不是一个固定的数值,对于不同核酸的测序反应,测序信号和校正信号的差异分布可能是不同的,需要针对每一次测序反应得到的差异分布确定阈值,再将大于阈值的测序信号舍弃。在
某些实现中,若截短后的序列的长度小于预设值,可以丢弃该整条序列,例如该数值可以是5,10,15,20,25,30,35,40,45,50,55,60,65,70,根据具体应用场景选择合适的数值。
[0242]
根据优选的实施方式,可以比较测序信号和校正信号的多种不同差异,综合判断后决定丢弃还是保留待筛选序列。例如,可以分别计算每个测序信号和校正信号的差值绝对值之和,以及每个测序信号和校正信号的差值平方值之和,根据上述两种差异做出差异分布图,将两种差异均较小的高质量信号保留,将两种差异均较大的低质量信号丢弃。
[0243]
本发明还提供一种核酸测序数据的筛选系统,其特征在于,包括:
[0244]
处理器,存储器,以及用于对核酸测序数据进行筛选的程序,所述程序包括如下指令:
[0245]
a)将待测核酸样品测序获得的测序信号进行信号校正,获得校正信号,所述校正信号可直接转换为碱基序列;
[0246]
b)对转换出的碱基序列,比较其在测序信号和校正信号中对应部分的信号差异;
[0247]
c)若测序信号和校正信号之间的所述信号差异大于给定的阈值,则丢弃该测序信号,否则予以保留。
[0248]
在本发明的核酸测序数据的筛选方法的具体实施部分中所讨论的特征中的每个特征同样适用于核酸测序数据的筛选系统的具体实施。如上所示,所有其他特征在此处不再重复,并且应被视为以引用方式重复。本领域普通技术人员将理解在这些具体实施中识别的特征可如何容易地与在其他具体实施中识别的基本特征组组合。
[0249]
实施例1
[0250]
从new england biolabs公司购买λ噬菌体的基因组dna,建库后进行纠错码测序。纠错码测序包含mk(两组测序底物分别是m(a,c),k(g,t))、ry(两组测序底物分别是r(a,g),y(c,t))、ws(两组测序底物分别是w(a,t),s(g,c))等3个round(一个round指的是用一种测序底物组合(如,mk)将待测序列全部测完的过程),设这3个round的测序中,一条原始荧光信号经信号归一化和失相校正后得到的信号(即测序信号s)分别为荧光信号经信号归一化和失相校正后得到的信号(即测序信号s)分别为经纠错校正后得到的信号(即校正信号c)分别为某个待评估质量的碱基在3个round的测序中分别位于cycle i、cycle j、cycle k,则定义该碱基的“失相校正信号与纠错校正信号间的差异”为:
[0251][0252]
利用bwa-mem软件将所得dna序列比对到参考基因组上,忽略未比对序列。根据比对结果,将凡是比对结果为“匹配”的碱基标记为“正确”,将比对结果为“错配”、“插入”或“缺失”的碱基标记为“错误”。将“失相校正信号与纠错校正信号间的差异”按数值大小划分为若干个区间,统计每个区间中碱基的准确率p,并按公式q=-10 log
10
(1-p)将其转化为质量值。“失相校正信号与纠错校正信号间的差异”与质量值的对应关系如图1所示,其中最高质量值达到43分,对应准确率99.995%。
[0253]
随后,针对另一份λ噬菌体的基因组dna进行纠错码测序。对测得的每个碱基,用同
样的方法计算失相校正信号与纠错校正信号间的差异,并根据图1所示的对应关系,评估出每个碱基的质量值。
[0254]
实施例2
[0255]
从new england biolabs公司购买λ噬菌体的基因组dna,建库后进行纠错码测序。对每个碱基,计算如下4种预测器的值:
[0256]
1.该碱基在序列中的位置;
[0257]
2.该碱基所处同源多聚物的长度;
[0258]
3.按实施例1的方法计算的失相校正信号与纠错校正信号间的差异;
[0259]
4.该碱基所处失相校正信号与其取整信号间的欧氏距离;
[0260]
利用bwa-mem软件将所得dna序列比对到参考基因组上,忽略未比对序列。根据比对结果,将凡是比对结果为“匹配”的碱基标记为“正确”,将比对结果为“错配”、“插入”或“缺失”的碱基标记为“错误”。利用上述4种预测器,通过phred算法构建质量评估表。该表共有447行,其中前20行如下:
[0261]
预测器1预测器2预测器3预测器4碱基质量值1384.50.60220.176444601.50.79430.1305451581.50.79430.1513441805.50.60220.1305441584.50.52070.1596441801.50.79430.130544405.50.60220.1678441804.50.69090.2101431181.50.92380.167843793.50.52070.2386431801.50.79430.1513431584.50.79430.1305431381.50.92380.1513431185.50.44060.1764431383.50.79430.1966421581.50.79430.1596431183.50.60220.2386421801.50.79430.1764421583.50.79430.1678421581.50.92380.151342
[0262]
利用该质量评估表对另一份λ噬菌体的基因组dna的纠错码测序数据进行评估。图2展示了评估结果中质量值在某一阈值以上的碱基的比例,其中质量值在40以上的碱基占55%,质量值最高达到45。图3展示了评估结果中打分的准确性,横坐标表示根据质量评估表预测出的质量值,纵坐标表示比对后实际测量出的质量值,圆盘大小表示碱基数量,虚线为方程y=x表示的直线,两条平行线为方程y=x
±
3表示的直线,它们围成的灰色平行四边
形为质量评估允许的波动范围。该结果表明所得质量评估表可准确评估碱基的测序质量。
[0263]
统计测序数据中质量值在40以上的碱基的替换错误(substitution)类型,如表2所示:
[0264]
表2
[0265][0266]
纠错码测序低至10-5
甚至10-6
数量级的错误率,使其在检测单核苷酸多态性(single-nucleotide polymorphism,snp)等基因变异上具备良好的性能。
[0267]
实施例3
[0268]
从new england biolabs公司购买λ噬菌体的基因组dna,建库后进行纠错码测序。对每个碱基,计算如下5种预测器的值:
[0269]
1.该碱基在序列中的位置;
[0270]
2.按实施例1的方法计算的失相校正信号与纠错校正信号间的差异;
[0271]
3.mk轮中该碱基所处简并多聚物中较多的一种碱基的数目;
[0272]
4.ry轮中该碱基所处简并多聚物中较多的一种碱基的数目;
[0273]
5.ws轮中该碱基所处简并多聚物中较多的一种碱基的数目。
[0274]
利用bwa-mem软件将所得dna序列比对到参考基因组上,忽略未比对序列。根据比对结果,将凡是比对结果为“匹配”的碱基标记为“正确”,将比对结果为“错配”、“插入”或“缺失”的碱基标记为“错误”。利用上述5种预测器,通过phred算法构建质量评估表。该表共有301行,其中前20行如下:
[0275]
[0276][0277]
该质量评估表的表现与实施例2中的质量评估表接近。
[0278]
实施例4
[0279]
从new england biolabs公司购买λ噬菌体的基因组dna样品,构建测序文库并进行纠错码测序,其中mk、ry、ws三轮均各反应50cycle,共采集到1797082个亮点具有完整的测序信号。其中,设每个亮点的测序信号中mk、ry、ws轮失相校正后的信号分别为测序信号。其中,设每个亮点的测序信号中mk、ry、ws轮失相校正后的信号分别为ecc校正后的信号分别为的信号分别为定义两种差异:
[0280][0281][0282]
其中对每个亮点的测序信号,均计算上述差异,其差异的分布如图4所示。显然,根据上述两种差异可将信号分为间隔明显的两簇,其中左下方一簇的两种差异均较小,为高质量信号,将予以保留;而右上方一簇的两种差异均较大,为低质量信号,将予以删除。据此完成高质量测序信号的筛选。
[0283]
以上仅为本发明较佳的实施方式,本发明所属领域的技术人员还能够对上述实施方式进行变更和修改,因此,本发明并不局限于上述的具体实施方式,凡是本领域技术人员在本发明的基础上所作的任何显而易见的改进、替换或变型均属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1