基于XGBoost模型结核病耐药基因变异识别方法
基于xgboost模型结核病耐药基因变异识别方法
技术领域
1.本发明涉及生物信息学、生物医学和机器学习领域,具体涉及xgboost模型、pfi算法和结核病耐药基因变异识别系统。
背景技术:
2.结核病(tb)仅次于艾滋病,是传染病死亡的主要原因。由于该疾病对全球结核病控制会造成威胁,因此,公共卫生组织也非常关切着耐药结核病的问题。 2014年,世界卫生组织(who)承认成功取决于制定更好的预防、诊断和治疗干预措施,并且决定使用20年的实验去消灭结核疾病。抗菌素耐药性在全球的出现构成了一项重大挑战。尽管呼吁普遍获得药物敏感性检测以指导个体化治疗,但检测费用高昂,且具备进行该检测所需技能的人员短缺,意味着在许多最需要的国家无法获得这种检测。
3.结核分枝杆菌(简称菌株)是结核病(tb)的病原体,菌株耐药性是结核病治疗过程中最主要的挑战之一。鉴于未经治疗的结核病有可能进一步传播,因此迫切需要加快为每个患者开出最佳药物的过程,以减少感染传播的风险。快速的分子诊断测试有助于确保及早发现并及时治疗,这些诊断的假设是只要存在一个之前确诊的单核苷酸多态性位点(snp),就足以引起耐药性。此类测试对引起药物耐药性的最常见突变有效,但是如果每种药物只有少量的目标点,这种方法就会受到限制。并且由于对某些抗结核药物的相关性了解甚少,鉴别这种多变量相关性的方法可能效用有限。
4.大多数技术都是基于每个药物的耐药变异库,但是,由于变异数据的高维性和未知的耐药机制,这些技术不一定会导致高分类性能,特别是对研究较少的药物。本次发明主要考虑到,一方面这些变异库大多是基于研究者们的研究需求所建立,需要更加丰富的数据,以研究更多耐药上的耐药基因变异识别;另一个方面要耐药性预测和和耐药基因变异识别技术需要进行改进。
5.因此,针对菌株耐药性预测,和结核病耐药基因变异识别的问题,使用了xgboost (extreme gradient boosting)模型提升了菌株耐药性预测的精确度,并且配合pfi(permutationfeature importance)算法识别耐药基因变异,以提高识别出的耐药基因变异的准确性。针对数据丰富的问题,本次研究使用的数据来源于由who认证的reseqtb(relational sequencingtuberculosis data platform)公开数据。近三年来,贡献者们向该平台上传了大量的菌株的基因变异信息和耐药性实验信息数据,推动了结核病领域的研究。相对于其他耐药变异库中的数据,该数据集变异基因多,变异更加丰富,是其他数据集的10倍以上。该平台上的数据结构较为繁琐,数据量较大,需要进一步的特征工程工作。
技术实现要素:
6.为克服现有技术的不足,本发明旨在预测发生基因变异的菌株对抗生素的耐药性,识别引起对抗生素耐药的重要基因变异。为此,本发明采取的技术方案是,基于xgboost模型结核病耐药基因变异识别方法,步骤如下:
7.(1)从公开数据源获取菌株基因变异数据和耐药性实验结果数据,其中,菌株基因变异数据中包含了菌株的基因变异状况,菌株与基因变异为多对多的对应关系;耐药性实验数据结果数据包含了菌株在抗生素药物上进行耐药性实验的结果,结果为耐药或者敏感,菌株与抗生素为多对多的关系;
8.(2)数据预处理:首先对耐药性实验结果数据进行处理,如果在对某一种抗生素进行耐药性实验的结果只有一种,则该药物对应的数据记录需要过滤掉,进一步滤掉掉没有实验结果以及有重复的实验记录数据;然后对菌株基因变异数据进行过滤,过滤掉核苷酸变化特征为空的样本和没有进行耐药性实验的菌株;
9.(3)构造模型数据集:对每一抗生素药物做一个数据集,用于对后续的每一种药物对应进行耐药性预测,和引起结核病耐药的重要基因变异特征识别。首先按照抗生素药物进行分组,每一组为菌株在该抗生素药物上,所有的耐药敏感实验结果;然后将把参与该药物耐药性实验的所有菌株的变异名称作为数据集特征,每一条样本表示一个菌株的所有基因变异信息,将每一种抗生素的实验结果和菌株变异信息通过菌株id联合在一起,即为每一种抗生素药物创建对应的基因变异耐药性数据集:
10.(4)模型训练,针对每种抗生素药在对应的数据集上使用xgboost模型做二分类训练,根据菌株的变异信息,预测该菌株是否耐药。在进行模型评估时,在每种抗生素药物所对应的数据集上都采用10次5折分层交叉验证的方法,使用auc(area under roc)指标对模型进行评估,结果取10次的平均值;
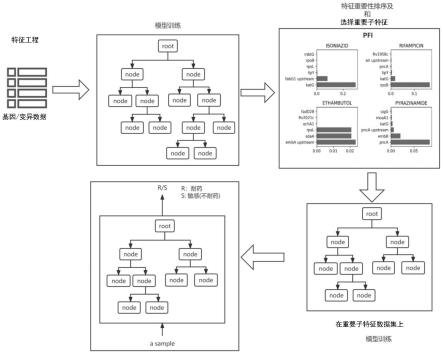
11.(5)耐药性基因变异识别:选用与模型无关的pfi算法,结合已经训练好的xgboost 模型,在各药物数据集上对基因变异特征进行重要性评估,按照重要性值对基因变异特征进行排序,其中,选取特征重要性值大于0.05的特征为对该药物发生抗药的重要基因变异特征,也为引起耐药的关键基因变异特征,将所选取的重要基因变异特征命名为重要子特征,对除重要子特征外的其他特征进行过滤后数据集命名为重要子特征数据集,这样得到的重要子特征为每一种抗生素所识别的耐药基因变异。
12.其中,步骤(5)选取特征重要性值大于0.01的基因变异特征,这些变异特征为所识别的引起该药物耐药的基因变异,变异所在的基因为所识别的引起该药物耐药的变异基因。
13.除此之外,还包括验证步骤,验证识别出的耐药基因变异的可靠性:将每种药物数据集根据所识别出的耐药变异特征进行筛选,数据集的特征只保留所识别的耐药变异特征,得到重要子特征数据集;然后,将每种抗生素药物对应的数据集训练新的xgboost模型,并且仍然采用10次5折分层交叉验证的方法使用auc指标对模型进行评估,与步骤(4)的评估结果进行对比。
14.本发明的特点及有益效果是:
15.本发明使用xgboost模型预测发生基因变异的菌株对抗生素的耐药性,然后结合pfi算法与已经训练好的xgboost模型,识别引起对抗生素耐药的重要基因变异。提高了菌株耐药性预测的精确度的同时,提高了结核病耐药基因变异识别的准确性。
附图说明:
16.图1本发明流程图。
具体实施方式
17.本发明涉及生物信息学、生物医学和机器学习领域,使用xgboost模型预测发生基因变异的菌株对抗生素的耐药性,然后与pfp算法结合识别引起结核病对抗生素耐药的重要基因变异。
18.菌株耐药性预测以及结核病耐药基因变异识别主要由三部分组成,分别为reseqtb数据的特征工程,菌株耐药性预测以及引起结核病耐药的重要基因变异的识别。这三个部分也是该领域研究面临的三个重要问题,为解决这些问题,本文给出了一套流程方法,步骤如下:
19.(1)从reseqtb上,获取菌株基因变异数据以及耐药性实验结果数据。其中,菌株基因变异数据中包含了菌株的变异状况,菌株与变异为多对多的对应关系。耐药性实验结果数据包含了菌株在抗生素药物上进行耐药性实验的结果,结果为耐药或者敏感,菌株与抗生素为多对多的关系。
20.(2)数据预处理。由于平台下载的数据不能直接用于机器学习模型,在模型训练前需要对菌株基因变异数据以及耐药性实验结果数据进行数据处理。首先对耐药性实验结果数据进行处理,如果在对某一种抗生素进行耐药性实验的结果只有一种,则该药物对应的数据记录需要过滤掉,然后滤掉没有实验结果以及有重复的数据记录。然后对菌株基因变异数据进行过滤,过滤掉基因变异特征为空的样本和没有参与耐药性实验的菌株。
21.(3)构造机器学习数据集。对每一抗生素药物做一个数据集,用于对后续的每一种药物对应一个模型进行耐药性预测,并且用于适应每一种药物的重要基因变异特征。首先,按照抗生素药物,对实验记录信息进行分组,每一组为一种抗生素药物上,菌株的耐药性实验结果。然后,将把参与该药物耐药性实验菌株的所有变异信息,作为数据集特征,每一条样本表示了一个菌株的所有变异信息。将每一种抗生素的实验结果和菌株变异信息通过菌株id联合在一起,即为每一种抗生素药物,创建对应的菌株耐药数据集。
22.(4)模型训练。本次研究使用了目前boosting算法中的最主流xbgoost模型,该算法在数据科学领域已经风生水起,但是在结核病耐药领域一直未被应用。本发明针对每种抗生素药,在对应的数据集上,使用xgboost模型做二分类训练,根据菌株上的所有变异信息,预测该菌株对该药物是否耐药。在进行模型评估时,在每种抗生素所对应的数据集上,都采用10次5折分层交叉验证的方法,并且使用auc指标对模型进行评估。采用多次交叉验证的目的是避免异常评估,使用auc指标主要考虑了结果数据集的不平衡性,该指标可以同时考虑正负样本。
23.(5)耐药性基因变异识别。探索引起抗生素耐药的重要基因变异,需要对基因变异特征按照某一种重要性指标进行特征重要性评估,重要性值较大的基因变异就是引起耐药性重要基因变异,也就是耐药性基因变异识别出的基因变异。由于xgboost模型在构造的过程中考虑的因素较多,因此用于评价特征重要的指标较多,使用不同的指标进行特征重要性排序会产生不同的结果。为解决这一问题,我们选用了与模型无关的pfi算法,对数据集中的变异特征进行特征重要性排序。得到排序结果后,选取特征重要性值大于0.05的特征为该药物抗药的重要基因变异特征,也为引起耐药的关键基因变异特征。将所选取的重要基因变异特征命名为重要子特征,对除重要子特征外的其他特征进行过滤后数据集命名为重要子特征数据集。这样得到的重要子特征为每一种抗生素所识别的耐药基因变异。专业
人员只需观察菌株上的变异是否在某一药物的重要子特征内,即可判断该菌株是否对该药物耐药。
24.(6)验证识别的耐药基因变异的可靠性。将在每种抗生素药物对应的重要子特征数据集上,训练新的xgboost模型,并且仍然采用10次5折分层交叉验证的方法使用auc指标对模型进行评估。与步骤(4)的评估结果进行对比,通过对比发现,在重要子特征数据集上训练出的模型,auc指标并没有下降,反而提升了2%,这也说明了对每种药物所识别的重要基因变异是可靠性是有保障的。每种药物上,新训练的xgboost模型,为我们得到的菌株耐药性预测模型和结核病耐药基因识别模型、
25.(1)数据集来源
26.本发明数据来源于reseqtb(https://platform.reseqtb.org),发布于2019年5月。该数据集是由两部分组成,一部分是菌株基因变异信息数据集,包括变异基因,变异位置,变异核甘酸等;另一部分是菌株耐药性实验数据集,包括实验药物信息,实验结果等。这个数据集通过菌株的id联系在一起。基因变异信息数据集包含了8025634条样本数据,每条样本包含了 31个特征,包括了菌株id,基因名字,变异类型,相关核苷酸,变异位置,核苷酸变化,相关序列等等,菌株id与核苷酸变化为多对多的关系。其中,菌株id为变异菌株的id信息,核苷酸变化特征是人类基因组变异协会(hgvs:human genome variation society)突变的命名法,此特征为菌株变异信息,基因名字特征为发生变异的基因,变异位置是相对于编码序列开始的核苷酸位置。变异类型只有核苷酸多态(snp)。菌株实验信息数据集包含了36376条样本数据,每一条样本数据有19个特征属性,包括了菌株id,耐药实验药物,耐性性结果等信息。该数据集包含了21种抗生素耐药的实验结果。四种一线药物的数量分别:isoniazid 药物5337株,rifampicin药物5110株,ethambutol药物4951株,pyrazinamide 药物4270株。实验结果分为三类,分别为中立(borderline),耐药(resistant)和敏感 (susceptible),其中敏感结果有24635条,耐药结果有7681条数据,中性结果有819条数据。
27.(2)数据预处理
28.基因变异信息数据集的特征工程如下:首先,明确本次研究的目的结菌株耐药性预测以及识别结核病耐药性重要基因及变异,根据实验目的选取至于本研究相关的数据特征,即只选取菌株id,基因名称和核苷酸变异三个特征用于后续实验。其次,通过统计发现,数据集中存在变异基因值以及核苷酸变异值为空现象,这些数据无法应用耐药性基因变异实验中,因此需要将这些数据去除。处理完成后的数据集还剩有6450494条样本数据,(每一个样本为菌株的一种变异),其中包含了6756个菌株在3617个基因上发生了228639种基因核苷酸变异。
29.实验数据集在本次研究中,第一步,由于本实验没有考虑不度下的药物进行研究,首先,我们需要去除不同的药物浓度下有相同的结果,将数据集中菌株id(isolate id),实验抗生素药物(msdrug)和耐药性结果(msstresc)三个特征数值都重复的样本去除,这也去除了不同浓度引起实验结果不同的数据,完成第一步数据去重后还剩下33135条样本数据。其次,实验数据中还存在845条不同浓度下有不同结果的实验数据,我们将这些实验数据去除,处理后的31443条数据就是不考虑不同浓度的实验数据。第二步,通过第一步的过滤后,我们发现特征msstresc值为borderline的数据只剩下56条,无法进行后续试验,我们将特征msstresc值为borderline的样本进行去除,第二步完成后,我们对数据集 msstresc特征
进行了数量统计,值为susceptible有23843条样本数据和值为 resistant有7544条样本数据。在前面提到,有四种药物只做了一次实验,只有一次实验数据,不能进行实验分析,我们将此4条数据进行删除。最终得到31383条样本数据,其中包括了4849个实验菌株。最后一步,进行特征提取,我们只提取菌株id(isolate id),实验抗生素药物(msdrug)和耐药性结果(msstresc)三个特征,形成处理后的实验信息数据集。
30.我们将两个数据集将特征菌株id作为为主键做内连接操作,这一操作既可以对菌株信息数据集和实验信息数据集两个数据集过滤,去除不能匹配的菌株,又可将核苷酸变异信息以及变异基因信息与实验结果关联起来。完成merge后得到了变异信息及其耐药性数据集,该新数据集由30319495条样本组成,其中包括了变异基因有3694个,核苷酸变异42282个。将数据集中msstresc特征中的文本特征转为机器学习模型可用的数值特征,数值特征特征列名为label,将resistant设置为1,susceptible设置为0。至此,得到的新数据集我们暂时称为变异信息及耐药性数据集,该数据的特征为菌株id(isolate id),基因名称 (genename),核苷酸变化(nucleotide change),实验抗生素药物(msdrug)和标签(label),处理后的数据集涵盖了所有菌株的所有核苷酸变异类型,变异的基因及其进行药物实验的结果。
31.(3)模型数据集构造
32.这对每一种抗生素药物,生产处所对应的实验数据集。数据集的步骤如下,首先将变异信息及耐药性数据集的特征菌株id(isolate id),核苷酸变化(nucleotide change),实验药物 (msdrug),标签(label)进行提取,然后根据实验药物(msdrug)特征进行分组,得到了每种药物的变异菌株以及实验结果,再根据菌株id(isolate id)进行分组,得到了每一个菌株的变异类别,并且将核苷酸变化(nucleotide change)特征了进行one-hot处理,即可得到每一种药物的变异数据集,该数据集的一条样本数据表示一个菌株该样本的特征为变异状况,样本的特征为所有菌株发生的核苷酸变异。若某一特征为1,表示菌株发生了该特征表示的变异,若某一特征为0,表示菌株没有发生该特征表示的变异。
33.(4)耐药性识别(特征选择)
34.结核病耐药基因变异识别最主要的工作是识别出引起结核病耐药的重要基因变异,也就是导致耐药的关键基因变异。识别重要基因变异的最核心工作是对数据集上的基因变异进行重要性评估,并且按照重要性得分进行特征排序,选取重要性值较大的基因变异特征。本次发明中,选用了与模型无关的pfi算法,该算法用于解释分类和回归模型,breima等人借助了随机森林中的思想,开发了一个与模型无关的特征重要性评估算法,它与分类和回归模型兼容。该算法的核心策略是考虑在模型训练完之后,将单个特征的数据值随机洗牌,破坏原有的对应关系后,再考察模型预测效果的变化情况,也就是说如果一个特性对预测结果没有用处,那么改变或排列它的值不会导致模型性能的显著降低。该算法评估基因变异特征重要性的具体流程为:1)将数据集第一列的特征列打乱2)然后将打乱后的数据集输入到已经训练好的xgboost模型中3)计算auc指标的下降值,该值为打乱基因变异特征重要值4)恢复到打乱前状态,将下一特征列打乱回到步骤2),直到求出每一个特征重要性。得到基因变异特征重要性后,对重要性值进行归一化处理,方便后续特征选择。
35.以上所述,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明披露的技术范围内,可轻易想到的变化或替换,都应
涵盖在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1