一种利用宏基因组分析溯源土壤耐药基因污染的方法
1.本发明涉及土壤生物学和生物信息学领域,具体是通过构建已知土壤微生物非冗余耐药基因集数据库,针对reads水平高通量注释宏基因组质控数据,实现土壤耐药基因污染的快速精准物种溯源。
背景技术:
2.农药、抗生素的滥用导致土壤中耐药基因(args)污染物的大量富集。一旦高风险的args传播给致病菌,或直接危害作物生长,或通过粮食蔬菜向食物链传播,严重威胁食物生产和人类健康。args检测和物种溯源对明确土壤args的风险等级至关重要。当前,args的检测已经相对成熟,如高通量pcr技术和宏基因组分析技术。然而,尽管高通量pcr技术靶向性强、应用广泛,但价格昂贵,检测结果受制于args特异性扩增引物的种类数量。宏基因组测序广谱性强、性价比高,可通过ardb、card、resfinder和sarg等数据库快速检测args的组成和丰度。然而,args的物种溯源刚刚起步,多依赖其他配套技术检测样品中的物种组成,再通过相关分析进行物种溯源。如高通量pcr技术依赖于扩增子测序技术,宏基因组分析技术依赖物种注释和contig组装等分析技术。
3.上述方法或过程繁琐,或基于blast比对费时费力、或阈值设置的主观性强,仅能实现简单笼统的物种溯源。因此,建立args物种溯源的宏基因组分析方法,为土壤耐药基因污染的溯源及其消减提供方法技术,具有重要意义。
技术实现要素:
4.本发明的目的是提供一种利用宏基因组分析溯源土壤耐药基因污染的方法,通过提取ncbi公共数据库中所有细菌参考基因组,创建非冗余基因集,比对ardb、card、resfinder和sarg的序列资源,构建了非冗余耐药基因集溯源数据库用于宏基因组测序分析,从而实现基于宏基因组分析溯源土壤args,为消减土壤耐药基因污染提供研究技术。
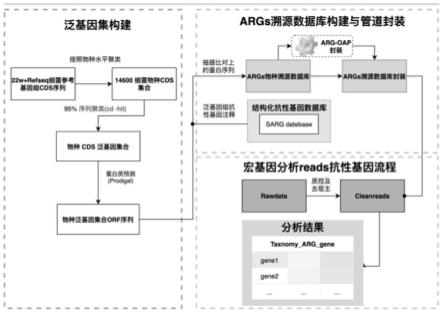
5.本发明采取的技术方案是:一种利用宏基因组分析溯源土壤耐药基因污染的方法,其包括以下步骤:
6.步骤一、构建非冗余耐药基因集溯源数据库;
7.步骤二、封装非冗余耐药基因集溯源数据库;
8.步骤三、土壤样品dna抽提和建库;
9.步骤四、采用illumina测序技术完成土壤样品的宏基因组测序;
10.步骤五、对测序数据进行质控并去除人类基因组污染,获取质控数据;
11.步骤六、对质控数据进行耐药基因定量分析,鉴定候选的耐药基因并获取基因的ppm、16s rrna以及细胞数量进行均一化后的丰度矩阵;
12.步骤七、不同样品的耐药基因丰度总体比较分析并可视化。
13.进一步的,所述步骤一中,数据库构建的步骤如下:
14.(1)从ncbi refseq数据库下载22w+参考基因组cds序列;
15.(2)按照物种水平合并所有cds序列;
16.(3)cds序列集合按照物种水平去冗余,构建出细菌物种的泛基因集合;
17.(4)对物种泛基因集合进行蛋白质预测;
18.(5)将预测的蛋白质序列与sarg数据库比对,抽提比对上的序列构建args物种溯源数据库。
19.进一步的,所述步骤二中,使用基于perl语言的args-oap软件封装非冗余耐药基因集溯源数据库。该方法与常规宏基因组分析思路不同,不需要拼接开放阅读框序列组装contigs。直接将宏基因组reads与args物种溯源数据库比对进行耐药基因的注释和定量,该方法不仅对土壤宏基因组测序信息中耐药基因的注释更为全面、定量更为准确,而且实现土壤耐药基因污染的物种溯源。
20.进一步的,所述步骤六中,样品的质控数据使用blastx比对args物种溯源数据库,获取抗性基计算每个基因每个样品的reads丰度;同时,在计算基因丰度的过程中,考虑不同基因参考序列长度的不一致,对丰度计算方法进行16s rrna的数目和细胞的数目均一化算法优化。
21.进一步的,通过usearch将质控序列比对copyrighter database 16s序列数据库,计算每个样本的比对上16s rrna的reads总数目及平均拷贝数,将16s rrna的reads总数除以16s rrna的平均拷贝数来估计细胞数,并对每个样品的抗性基因的丰度进行16s reads数目和细胞数量均一化;最终的结果具有3种丰度定量单位,分别为每百万条reads中匹配基因的reads数目(ppm)、每个16s rrna基因匹配基因的拷贝数和每个细菌中匹配基因的拷贝数。
22.更进一步的,细胞数的计算公式如下:
[0023][0024]cnumber
表示每个样品的细胞数量,n
16sseq
是比对到16s数据库的reads数量,l
reads
表示reads长度,l
16sseq
表示16s长度,ai表示样品比对到高可变区对齐后分类学单元的数量,a是所有m个高可变区对齐后的分类学单元总数,mi表示copyrighter数据库内分类学单元的拷贝数量。
[0025]
更进一步的,耐药基因丰度计算公式为:
[0026][0027]
其中abundance
16s
表示16s标准化耐药基因丰度,16si表示样品比对到16s序列的reads数量,16s
len
表示16s的序列长度,n
gene
表示耐药基因的reads丰度,l
gene
表示耐药基因长度;
[0028][0029]
其中abundance
cell
表示细胞数标准化耐药基因丰度,16si表示样品比对到每个16s序列的reads数量,16s
len
表示16s的序列长度,n
gene
表示耐药基因的reads丰度,l
gene
表示耐
药基因长度。c
number
表示每个样品的细胞数量。
[0030]
与现有技术相比,本发明的有益效果如下:
[0031]
(1)目前宏基因组序列args物种溯源难以实现,现有的方法还是基于contig组装序列进行args注释,提取比对上数据库的args序列后再比对nr数据库进行物种注释,步骤过于复杂且溯源精确度较低。而本发明构建args物种溯源数据库中的args序列已经具备微生物物种信息注释,从根源上解决宏基因组在进行args注释定量的时候物种溯源的问题。
[0032]
(2)本发明成功构建细菌物种水平的非冗余基因集,并实现了对14600个细菌物种进行args注释,为全球微生物args图谱鉴定及分析提供了一个新的角度。
[0033]
(3)所构建的args物种溯源数据库具有很好的延展性,不仅可以用于宏基因组质控数据的args物种溯源定量分析,对于其他高通量测序所获得的数据(如宏转录组,细菌扫描及完成图等)同样都可以使用该数据库进行args注释;且由于数据库args序列本身具有物种信息,对微生物基因组序列进行args注释时甚至可以评估其所携带的args宿主来源,为抗性基因水平基因组转移等相关研究提供了技术支撑。
[0034]
本发明的附加方面和优点将在下面的描述中部分给出,部分将从下面的描述中变得明显,或通过本发明的实践了解到。
附图说明
[0035]
图1是args物种溯源分析流程图;
[0036]
图2是refseq物种非冗余基因集基因组数量分布直方图;
[0037]
图3是样品中耐药基因物种溯源丰度表图(以ppm为例);
[0038]
图4是args物种溯源注释携带args物种相对丰度(genus水平);
[0039]
图5是args物种溯源注释args种类相对丰度(args type水平)。
具体实施方式
[0040]
下面通过实施例对本发明进行具体的描述,文中未注明具体条件的实验方法,通常按照常规条件或按照仪器制造商所建议的条件进行。除非另行定义,文中所使用的所有专业与科学用语与本领域技术人员所熟悉的意义相同。
[0041]
如图1和2,本发明提供了一种利用宏基因组分析溯源土壤耐药基因污染的方法,包括以下步骤:
[0042]
步骤一、下载参考基因组cds序列构建物种非冗余基因集;
[0043]
步骤二、非冗余基因集比对sarg数据库构建非冗余耐药基因集溯源数据库;
[0044]
步骤三、perl语言封装数据库,搭建宏基因组args物种溯源的分析流程;
[0045]
步骤四、土壤样品dna抽提和建库;
[0046]
步骤五、采用illumina测序技术完成土壤样品的宏基因组测序;
[0047]
步骤六、对测序数据进行质控并去除人类基因组污染,获取质控数据;
[0048]
步骤七、质控数据输入宏基因组args物种溯源的分析流程,鉴定候选的耐药基因并获取基因的ppm、16s rrna以及细胞数量进行均一化后的丰度矩阵;
[0049]
步骤八、不同样品的耐药基因丰度总体比较分析并可视化。
[0050]
关于非冗余耐药基因集溯源数据库构建:
[0051]
下载ncbirefseq数据库中22万个参考基因组的编码蛋白(cds),使用cd-hit软件构建物种非冗余基因集(参数:-n 9-g 1-c 0.95-g 0-m 0-d 0-as 0.9),囊括14600个物种。经blastp严格比对(参数:》=95%similarity and》=85%gene coverage)筛选物种非冗余基因集,获取耐药基因的物种信息,最终建立包含24930条耐药基因的非冗余耐药基因集溯源数据库,简称args物种溯源数据库。数据库构建的具体步骤如下:
[0052]
步骤1:ncbi refseq数据库下载22w+参考基因组cds序列;
[0053]
步骤2:按照物种(species)水平合并所有cds序列;
[0054]
步骤3:cds序列集合按照物种水平去冗余,构建出14600个细菌物种(cds)的泛基因集合;
[0055]
步骤4:对物种泛基因集合进行蛋白质预测;
[0056]
步骤5:将预测的蛋白质序列与sarg数据库比对,抽提比对上的序列构建args物种溯源数据库。
[0057]
关于非冗余耐药基因集溯源数据库封装:
[0058]
使用基于perl语言的args-oap软件封装args物种溯源数据库,建立耐药基因溯源的宏基因组分析新方法。该方法与常规宏基因组分析思路不同,不需要拼接开放阅读框序列组装contigs。直接将宏基因组reads与args物种溯源数据库比对进行耐药基因的注释和定量,该方法不仅对土壤宏基因组测序信息中耐药基因的注释更为全面、定量更为准确,而且实现土壤耐药基因污染的物种溯源。
[0059]
关于宏基因组质控数据进行耐药基因定量分析:
[0060]
args-oap软件内部已经通过的usearch+blastx结合的方式来比对args物种溯源数据库的方式对宏基因组测序reads进行比对和定量。
[0061]
具体来说:所有样品的质控数据使用blastx比对args物种溯源数据库,获取抗性基计算每个基因每个样品的reads丰度。同时,在计算基因丰度的过程中,考虑了不同基因参考序列长度不一致的问题,对丰度计算方法进行了16s rrna的数目和细胞的数目均一化算法优化。通过usearch将质控序列比对copyrighter database 16s序列数据库,计算每个样本的比对上16s rrna的reads总数目及平均拷贝数,将16s rrna的reads总数除以16s rrna的平均拷贝数来估计细胞数(见公式2),并基于公式1和3对每个样品的抗性基因的丰度进行16s reads数目和细胞数量均一化。因此,最终的结果具有3种丰度定量单位,分别为每百万条reads中匹配基因的reads数目(ppm)、每个16s rrna基因匹配基因的拷贝数和每个细菌中匹配基因的拷贝数。
[0062]
16s均一化的耐药基因丰度公式:
[0063][0064]
其中abundance表示耐药基因丰度,16si表示样品比对到16s序列的reads数量,16s
len
表示16s的序列长度,n
gene
表示耐药基因的reads丰度,l
gene
表示耐药基因长度。
[0065]
细胞均一化的耐药基因丰度公式:
[0066][0067]cnumber
表示每个样品的细胞数量,n
16sseq
是比对到16s数据库的reads数量,l
reads
表
示reads长度,l
16sseq
表示16s长度,ai表示样品比对到高可变区对齐后分类学单元的数量,a是所有m个高可变区对齐后的分类学单元总数,mi表示copyrighter数据库内分类学单元的拷贝数量。
[0068][0069]
其中abundance
cell
表示耐药基因丰度,16si表示样品比对到每个16s序列的reads数量,16s
len
表示16s的序列长度,n
gene
表示耐药基因的reads丰度,l
gene
表示耐药基因长度。c
number
表示每个样品的细胞数量。
[0070]
下面结合具体实例作进一步说明:
[0071]
本实施例使用来自全国17个地区采集的土壤进行盆栽试验,每个地区的土壤分别执行ck(对照组)与rs(抗生素处理组),实施抗性基因物种溯源检测和定量的新方法,包括以下具体步骤:
[0072]
样品dna抽提和建库,采用illumina测序技术完成环境样品的宏基因组测序,对测序数据进行质控并去除人类基因组污染。
[0073]
使用args-oap工具封装非冗余耐药基因集溯源数据库后,直接对质控后的clean reads分别进行耐药基因物种溯源注释和定量,与参考数据库进行比对时,比对参数设置期望值e-value为1e-7,相似性》=80%,相似的氨基酸长度》=25aa。
[0074]
将质控后的reads比对16s rrna数据库计算每个样本的比对上16s rrna的reads总数目和细胞数量,再使用每个样品16s reads总数目和细胞数量对每个样品每个耐药基因的丰度进行均一化,耐药基因丰度见图3。args物种溯源表中的args组成以及携带args的物种组成的相对丰度分别如下图4,5。
[0075]
以上显示和描述了本发明的基本原理、主要特征和优点。本领域的普通技术人员应该了解,上述实施例不以任何形式限制本发明的保护范围,凡采用等同替换等方式所获得的技术方案,均落于本发明的保护范围内。
[0076]
本发明未涉及部分均与现有技术相同或可采用现有技术加以实现。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1