基于血管生成相关基因的肿瘤预后预测模型构建方法
1.本发明涉及生物信息领域,具体地说,是基于血管生成相关基因的肿瘤预后预测模型构建方法。
背景技术:
2.胃癌(gc)是全球第五大最常见的恶性肿瘤,也是第三大最常见的癌症死亡原因,尽管治疗策略取得了巨大进展,但其结果仍然令人沮丧。本质上,gc是一种高度异质性疾病,具有不同的特征,包括位置、组织学类型、分子分类和生物学行为。然而,传统的诊断方法主要基于肿瘤、淋巴结、转移(tnm)分期,容易忽略具有生物学异质性的原发肿瘤。因此,构建一个风险模型来识别高危患者并为gc患者的个性化治疗提供指导至关重要。
3.血管生成被定义为新毛细血管从已有血管生长的过程,并被认为是多发性实体瘤中肿瘤细胞增殖、生长和转移的重要因素。它被认为是癌症的标志之一,因为它通过提供氧气和营养物质、传递赋予免疫和治疗抵抗力的分子以及促进癌细胞转移的能力来促进癌细胞的生长。之前的研究表明,多种基因在血管生成中起着关键作用,如血管内皮生长因子(vegfs)及其受体(vegfrs),这些基因在血管生成信号通路中得到了很好的研究。之前的研究表明,在gc中,活跃的血管生成可导致治疗失败和不良结果(g.roviello,r.petrioli,l.marano,k.polom,d.marrelli,a.perrella,and f.roviello,angiogenesis inhibitors in gastric and gastroesophageal junction cancer.gastric cancer 19(2016)31-41.)。feng等人发现,在免疫生态位高度浸润的gc患者中,血管生成信号与细胞毒性功能显著相关(f.yi,y.dai,z.gong,j.n.cheng,b.j.o.zhu,and therapy,association between angiogenesis and cytotoxic signatures in the tumor microenvironment of gastric cancer.volume 11(2018)2725-2733.)。然而,gc靶向血管生成的临床相关性尚未得到充分评估。
4.癌相关成纤维细胞(caf)是肿瘤微环境(tme)的主要组成部分,并参与肿瘤微环境中的关键过程,包括细胞外基质(ecm)重塑、与癌细胞的相互信号交互作用以及与浸润性炎性细胞的串扰。越来越多的证据表明,caf在肿瘤的生长、侵袭和进展中起着至关重要的作用(d.dayan,t.salo,s.salo,p.nyberg,s.nurmenniemi,d.e.costea,and m.vered,molecular crosstalk between cancer cells and tumor microenvironment components suggests potential targets for new therapeutic approaches in mobile tongue cancer.cancer med 1(2012)128-40.c.j.hanley,m.massimiliano,f.kirsty,s.m.thirdborough,m.toby,s.j.frampton,d.m.smith,h.elena,s.cedric,and b.j.j.n.c.i.marc,targeting the myofibroblastic cancer-associated fibroblast phenotype through inhibition of nox4.(2018)1.l.liu,l.liu,h.h.yao,z.q.zhu,and q.j.p.o.huang,stromal myofibroblasts are associated with poor prognosis in solid cancers:a meta-analysis of published studies.11(2016)e0159947.)。此外,最近的研究表明,血管生成在caf促进肿瘤侵袭和转移中起着关键作用(d.tang,j.gao,
s.wang,n.ye,y.chong,y.huang,j.wang,b.li,w.yin,and d.j.t.b.wang,cancer-associated fibroblasts promote angiogenesis in gastric cancer through galectin-1expression.37(2016)1889-1899.)。然而,与血管生成相互作用的caf与gc患者预后之间的相关性尚未明确。
技术实现要素:
5.本发明的目的在于提供基于血管生成相关基因的肿瘤预后预测模型构建方法。
6.本发明从癌症基因组图谱(tcga)下载了414例包含临床特征和rna-seq数据的gc患者,并对血管生成相关基因的表达进行了相关性分析。单样本基因集富集分析(ssgsea)用于分析gc患者的args特征。此外,本发明还调查了arg和cafs含量之间的相关性,以及它们对gc患者生存率的影响。然后,args亚型预测模型可以在gc患者中有力地区分这两种亚型,该预测模型由多个机器学习算法(mmla)构建,并使用两个独立的gc队列进行验证。通过分析args评分高或低的gc患者在基因组特征、tme、免疫治疗疗效和靶向治疗方面的差异,本发明发现该预测模型在确定患者预后和治疗目标方面具有很好的指导意义。
7.本发明的第一方面,提供基于血管生成相关基因args的肿瘤预后预测模型构建方法,包括以下步骤:
8.步骤s1:从tcga某癌种队列中收集原始rna测序(rna seq)数据集和临床特征,随机分为训练集和测试集;
9.步骤s2:从分子签名数据库(http://www.gsea-msigdb.org/gsea/msigdb/,msigdb-hallmark版本7.4)下载具有表达谱的args基因集;
10.步骤s3:在训练集中,通过计算每个变量的重要性分数(应用glmnet、rms、e1071、caret、randomforest、boruta,以及r中的xgboost软件包来实现),使用支持向量机(svm)、最小绝对收缩和选择算子(lasso)回归、随机森林和boruta(rfb)以及极端梯度增强(xgboost)分析,选择最重要的组相关特征;
11.选择args相关差异表达基因的表达作为输入变量(自变量),选择亚型状态作为结果(二元因变量,0或1);使用受试者工作特征(roc)曲线来评估训练集中用于特征选择的四种机器学习算法的性能,并随后比较roc曲线(aucs)下的区域;然后,从lasso、svm、rfb和xgboost分析中的交叉基因中获得最关键的亚型相关基因,并使用venn图进行可视化;最后,对关键基因进行多元logistic回归分析,并将其用于构建预测模型;roc曲线用于研究亚型预测因子的性能,进而确定区分不同亚型的最佳截止值,以及auc、敏感性、特异性和准确性;最后,以同样的方式使用测试集验证预测模型的预测性能。
12.进一步的,所述的肿瘤包括但不限于胃癌、膀胱癌。
13.本发明在证明了胃癌arg评分与预后相关的基础上,分别用lasso、rf、svm和xgboost四种方法进行测序候选基因,本发明保护所有这些方法做得出的基因组合,即degs.lasso,rf,svm,and xgboost分别分析出的33,114,80and 110基因。
14.原则上4种不同的方法都可以,可以产生四种不同的公式,甚至两两都可以,三三可以,有20多种公式基因方式。但无疑,这种4种方法组合起来的才是最准确的。最终选定四种方法共有的10个基因作为预测模型的基因组成。
15.本发明的第二方面,提供一种肿瘤预后预测模型,其通过如上所述的方法建立。
16.进一步的,所述的肿瘤预后预测模型的计算公式如下:
17.预测分值=5.869+0.852
×
(dclk1的表达水平)+0.295
×
(ptgis的表达水平)+0.340
×
(nudt10的表达水平)+0.598
×
(zfhx4的表达水平)+0.290
×
(pcdh9的表达水平)+0.211
×
(chrdl1的表达水平)+0.073
×
(nlgn1的表达水平)+0.298
×
(agtr1的表达水平)+0.221
×
(cntn1表达水平)+0.261
×
(ecrg4表达水平)。
18.进一步的,所述的表达水平包括但不限于基因表达水平、基因的mrna表达水平、基因编码蛋白的表达水平。
19.进一步的,所述基因的mrna表达水平的测定方法包括pcr、基因芯片、二代高通量测序、panomics或nanostring宏基因组测序中的任意一种或至少两种的组合。
20.这种模型可以用于不同的肿瘤病人群体,即对于一个新的群体,可以通过增加标本量,计算“预测分值”,找到中间值即介值,在此基础上对后续的患者进行打分和进行分型。本发明保护应用该公式对不同群体进行检测计算并最终确定值。
21.本发明优点在于:
22.1、本发明共鉴定出36种args表达水平与gc患者的无病生存率(dfs)和总生存率(os)密切相关。根据他们的表达,ssgsea建立了arg相关的预后特征,在tcga队列中,预测1年、3年、5年os和dfs的曲线下面积(auc)分别为0.61、0.64、0.76和0.61、0.68、0.65。三个独立的地理数据集(包括gse26253、gse26091和gse66229)进一步验证了该特征的预测能力。此外,通过spearman分析,args信号与癌症相关成纤维细胞(caf)显著相关,同时具有高args和基质caf特征的gc患者的生存率最差。应用多重机器学习算法(mmla)进一步建立10基因arg亚型预测模型,并使用两个外部独立gc队列进一步验证。args预测模型的得分分值高较分值低的预后差,预测分值得分高与免疫治疗和潜在抗her2或fgfr4治疗的疗效较低相关,但对抗血管生成相关治疗更敏感。总之,本发明的结果表明,新的基于args的分类为gc提供了一个非常有希望的预后预测模型,并可作为医生选择优先使用免疫治疗和靶向治疗的潜在应答者的指南。
23.2、本发明提供肿瘤预后相关基因及其在制备肿瘤预后预测诊断产品中的应用,本发明开发的肿瘤预后风险预测系统对无病生存率(dfs)和总生存率(os)的判别能力明显优于其他研究报道的基因评分系统。此系统可用于辅助预测肿瘤患者对治疗干预的反应,进行免疫治疗或靶向治疗方案选择,达成个体化医疗的目的。
24.3、本发明证实了caf与gc血管生成过程相关基因表达的相关性。此外,在本发明中,使用gc将患者分为ⅰ型亚型和ⅱ型亚型组,并预测不同亚型患者对免疫治疗的反应。这些结果为未来选择免疫治疗患者提供了一种潜在的方法。它们可以作为预测gc患者预后和诊断的潜在生物标志物发挥重要作用,并为gc患者的靶向治疗提供新的视角。
附图说明
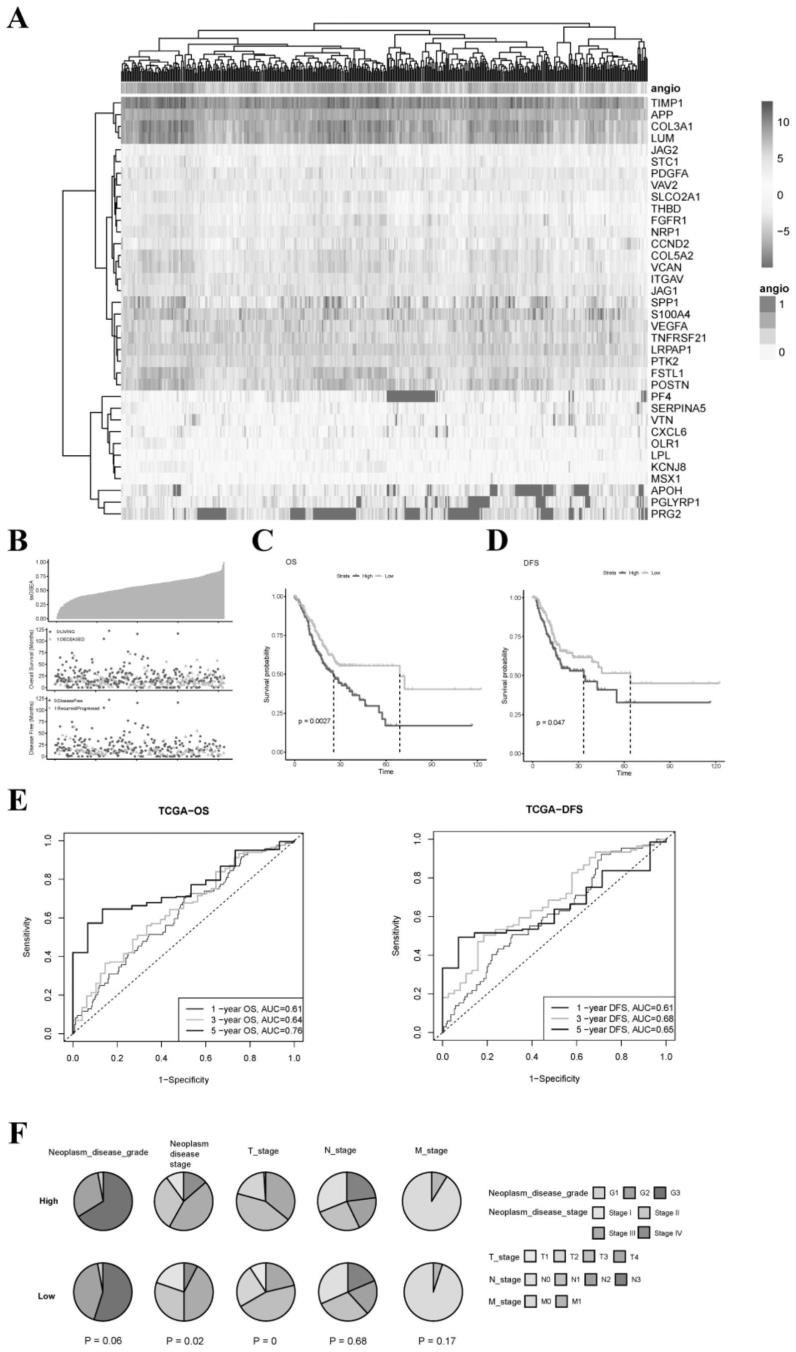
25.图1:tcga队列中候选血管生成相关基因的识别和预后分析。a:36个血管生成相关基因在胃癌组织中表达的热图。b:tcga队列中ssgsea得分的分布和中值(up面板)。tcga队列中os、os状态和风险评分的分布(中间面板)。tcga队列中dfs的分布、dfs状态和风险评分。c:tcga队列中患者os的kaplan
–
meier曲线,该队列分为高危组和低危组。d:tcga队列中患者dfs的kaplan
–
meier曲线,该队列分为高危组和低危组。e:roc(受试者操作特征)曲线,
显示tcga队列中args信号的1年、3年和5年os和dfs预测效率。f:gc队列中高args预后评分组和低args预后评分组之间的临床特征分析。
26.图2:胃癌患者arg与生存率之间的相关性分析。a:胃癌患者总体和无病生存率的独立预后因素。tcga数据集中患者临床病理特征(包括风险评分)与总生存率和无病生存率之间关系的单变量和多变量cox回归分析。b:在tcga队列(包括i期和ii期、ii期和iii期、g1-2期和g3期)中,对胃癌患者ssgsea评分与生存率(os和dfs)之间的相关性进行分层分析。t1-2和t3-4,无阶段和n1-3阶段,m0阶段和m1阶段。
27.图3:gc队列中高args标记分数组和低args标记分数组的tme。a:ssgsea评分与icg、cyt、hla、估计值、til之间的斯皮尔曼秩次相关。b:ssgsea评分与纯度的相关性分析。c和d:比较高风险组和低风险组的免疫细胞丰度。
28.图4:不同arg和caf亚组的生存分析。tcga队列中患者os和dfs的kaplan
–
meier曲线分为高和低caf密度组(a、b)、mcaf(c、d)、vcaf(e、f)、dcaf(g、h)。arg和caf之间的斯皮尔曼秩次相关性包括总caf(i,r=0.84,p=2.2e-16)、mcaf(j,r=0.64,p=8e-14)、vcaf(k,r=0.31,p=0.00089)、dcaf(l,r=0.52,p=5.4e-09)。根据args特征评分和mcafs内容,os(m)和dfs(n)的kaplan
–
meir曲线。caf:癌症相关成纤维细胞;血管性caf:vcaf;mcaf:matrix-caf;dcaf:发育性caf。
29.图5:三个验证集中arg特征风险评分的预后表现。kaplan
–
gse26901(a)、gse26253(b)和gse96229(c)队列的总生存率和无病生存率的meier曲线。
30.图6:args亚型预测因子的构建和验证。a:tcga队列中两个集群(集群1和集群2)的主成分分析(pca)。b:不同簇的不同表达基因的维恩图。tcga队列中患者os(c)和dfs(d)的kaplan
–
meier曲线,该队列分为第1组和第2组。e:第一组和第二组之间免疫评分、基质评分和评估评分的比较。f:tim3在两个簇中的表达水平。通过估算(g)和xcell(h)比较第1组和第2组之间浸润性免疫细胞的含量。通过venn图确定了10个最关键的亚型特异性基因,这些基因由四种特征选择算法共享。在列车组(j)和测试组(k)中,区分两个亚型时,亚型预测因子的roc曲线。kaplan
–
meier生存分析还表明,在gse26901(l-m)队列中,i型和ii型患者的os和pfs显著不同。roc曲线显示gse26901队列中args信号的1年、3年和5年os(j)和dfs(k)预测效率。kaplan
–
meier生存分析还表明,在gse66229队列(p-q)中,i型和ii型患者的os和pfs显著不同。j-k:roc曲线显示gse66229队列中args信号的1年、3年和5年os(r)和dfs(s)预测效率。roc:接收器工作特性。os:总体生存率;dfs:无病生存率。
31.图7:通过args分类识别分子特征,以及args评分与抗pd-l1免疫治疗之间的关联分析。答:gc患者复发率的比较。b:比较gse66229中四种不同的arg类型,包括msi、tp53阳性、tp53阴性和emt。c:不同tcga-stad分子亚型之间的预测差异。方框的上端和下端代表四分位值的范围。框中的线条代表中值。kruskal-wallis检验用于比较五种tcga-stad分子亚型之间的统计差异(p《0.0001)。d-e:由args评分高(d)和args评分低(e)的人建立的肿瘤体细胞突变瀑布图。每一列都表示个别患者。上面的条形图显示tmb,右边的数字表示每个基因的突变频率。右边的条形图显示了每种变异类型的比例。f:imvigor210队列中根据args评分亚型得出的总生存率kaplan
–
meier曲线。g:imvigor210队列中抗pd-l1免疫治疗反应不同的患者args评分分布的箱线图。显著性由wilcoxon检验确定。sd,病情稳定;进展性疾病;cr,完全反应;pr,,部分反应。h:突变计数、fgfr4、her2和args评分之间的相关性。i:高
风险或低风险评分样本之间舒尼替尼、索拉非尼、帕佐帕尼和阿西替尼的ic50差异。ic50:半抑制浓度。
32.图8:(a)tcga-gc队列中36个血管生成相关基因的基因组改变。左图:tcga-gc队列中36个血管生成相关基因(arg)的基因组改变;高(中面板)和低arg预后评分组(右面板)中前20个流行基因的遗传改变。args mrna与拷贝数变异(b)和甲基化水平(c)之间的相关性;(d)有或没有单个精氨酸基因组改变的样本之间精氨酸预后评分的差异;(e)高风险组和低风险组中每个被调查arg拷贝数的差异;(f)高风险组和低风险组中每个被研究的精氨酸甲基化水平的差异。*p《0.05;**p《0.01;ns:不重要。
33.图9:三个验证队列中gc患者cafs亚型与arg信号之间的关联。gse26901(a)、gse66229(b)和gse26253(c)中高arg风险评分和低arg风险评分之间的生存率差异。(d)args与基质癌相关成纤维细胞(mcafs)的相关性。os:总体生存率;dfs:无病生存率;rfs:复发。
34.图10:gse26253中args标记相关肿瘤微环境分析。(a)gse26253中高args评分和低args评分样本间基质、免疫和评估评分的差异。(b)采用spearman分析法分析ssgsea评分与tilss、icgs(ctla4、tim3、lag3和ido1)之间的相关性。通过xcell(c)和epic(d)分析肿瘤浸润的免疫细胞p《0.05;**p《0.01;***p《0.001;****p《0.0001;ns:不重要。
35.图11:gse26901中args标记相关肿瘤微环境分析。(a)gse26901中高args评分和低args评分样本间基质、免疫和评估评分的差异(b)ssgsea评分与tilss、icgs(ctla4、tim3、lag3和ido1)之间的相关性通过spearman分析进行分析。通过xcell(c)和epic(d)分析肿瘤浸润的免疫细胞p《0.05;**p《0.01;***p《0.001;****p《0.0001;ns:不重要。
36.图12:gse66229中args标记相关肿瘤微环境分析。(a)gse66229中高args评分和低args评分样本间基质、免疫和评估评分的差异。(b)采用spearman分析法分析ssgsea评分与tilss、icgs(ctla4、tim3、lag3和ido1)之间的相关性。通过xcell(c)和epic(d)分析肿瘤浸润的免疫细胞p《0.05;**p《0.01;***p《0.001;****p《0.0001;ns:不重要。
具体实施方式
37.下面结合实施例对本发明提供的具体实施方式作详细说明。
38.实施例:
39.1.方法和材料
40.1.1胃癌数据集的获取和血管生成相关基因的选择
41.tcga胃癌队列的原始rna测序(rna seq)数据集和临床特征可从tcga网站下载(http://xena.ucsc.edu/)作为培训组。gse26253(432个gc样本)、gse26901(109个gc样本)和gse96229(185个gc样本)从基因表达综合(geo,https://www.ncbi.nlm.nih.gov/geo/)用于验证研究。
42.从分子签名数据库下载血管生成相关基因共36个(http://www.gsea-msigdb.org/gsea/msigdb/,msigdb-hallmark版本7.4),作为args基因集。
43.1.2基于单样本基因集富集分析(ssgsea)的args“signature”构建
44.根据给定的args表达谱,通过经验累积分布函数,使用ssgsea计算富集分数,即每个样本中一个基因集的绝对富集程度。使用基因集变异分析(gsva)软件包计算了来自
tcga-stad的414个样本的args ssgsea得分。r.的limma软件包用于评估不同组织类型和生存状态的评分。gsea(版本4.1.0.24)用于对基因集(血管生成相关基因)进行基因集富集分析(gsea),使用414个肿瘤样本的基因表达谱进行1000个排列。将患者分为高ssgsea评分组和低ssgsea评分组进行进一步分析。
45.1.3args的差异表达分析和遗传改变
46.为了进一步探索每个血管生成相关基因的潜在作用,这些基因将与特征相符,使用cbioportal提取args数据,以证明tcga-gc队列中gc患者的每个基因突变(错义、剪接、截断、扩增和深度缺失改变等)。此外,甲基化和拷贝数变异(cnv)来自cbioportal数据库。spearman分析用于研究甲基化、cnv和arg表达之间的相关性。
47.1.4肿瘤浸润免疫细胞分析
48.我们首先评估了cyt、estimate、hla、icg、til、肿瘤纯度和arg评分之间的相关性。根据“epic和xcell”软件包,使用epic和xcell方法计算tcga和geo数据集中免疫细胞的浸润丰度,其中epic软件包(版本1.1.5)中有8个免疫细胞,xcell软件包中有36个免疫细胞(https://xcell.ucsf.edu/)分别为。
49.1.5通过多种机器学习方法构建和验证args亚型预测因子
50.414名gc患者按4:1的比例分为训练组(n=331)和测试组(n=83)。首先,在训练集中,使用支持向量机(svm)、最小绝对收缩和选择算子(lasso)回归、随机森林和boruta(rfb)以及极端梯度增强(xgboost)分析,通过glmnet、rms、e1071、caret、randomforest、boruta,以及r(w.sauerbrei,p.royston,and h.j.s.i.m.binder,selection of important variables and determination of functional form for continuous predictors in multivariable model building.26(2010)5512-5528.margin-maximised redundancy-minimised svm-rfe for diagnostic classification of mammograms,int j data min bioinform,2014,pp.374.j.yperman,t.becker,d.valkenborg,v.popescu,and l.m.peeters,machine learning analysis of motor evoked potential time series to predict disability progression in multiple sclerosis.(2019).w.li,y.yin,x.quan,and h.j.f.i.g.zhang,gene expression value prediction based on xgboost algorithm.10(2019)1077-.)中的xgboost软件包。选择args相关差异表达基因的表达作为输入变量(自变量),选择亚型状态作为结果(二元因变量,0或1)。使用受试者工作特征(roc)曲线来评估训练集中用于特征选择的四种机器学习算法的性能,并随后比较roc曲线(aucs)下的区域。然后,从lasso、svm、rfb和xgboost分析中的交叉基因中获得最关键的亚型相关基因,并使用venn图进行可视化。最后,对关键基因进行多元logistic回归分析,并将其用于构建被称为“亚型预测因子”的预测模型。roc曲线用于研究亚型预测因子的性能,进而确定区分不同亚型的最佳截止值,以及auc、敏感性、特异性和准确性。最后,使用测试集来验证亚型预测因子的预测性能也以类似的方式进行。
51.1.6抗血管生成治疗反应的疗效评估
52.来自癌症药物敏感性基因组学(gdsc,https://www.cancerrxgene.org/)下载这些研究以探索高风险组和低风险组之间抗血管生成治疗方案的反应。以半数最大抑制浓度(ic50)作为疗效评价指标。
53.1.7统计分析
54.所有统计分析和绘图均通过r软件(3.6.0版)进行。风险评分与临床特征的相关性采用x2检验。绘制kaplan
–
meier曲线,采用对数秩检验检验各组间os的显著性差异。单变量和多变量cox比例风险回归分析也用于评估风险评分与os之间的关系。roc分析用于检测基因特征风险评分预测生存率的敏感性和特异性。roc曲线下面积(auc)可作为判断预后准确性的指标。在所有分析中,p值《0.05被设定为具有统计学意义。
55.2.结果
56.2.1 tcga队列中候选血管生成相关基因的识别和预后分析。
57.研究gc中与血管生成相关的基因变异。首先,在tcga队列中总共选择了36个具有表达谱的arg(图1a)。根据ssgsea得分的中值,tcga队列中的所有gc患者被分为高危组和低危组(图1b,上图)。图1b中的中间和底部显示,高危组患者的生存率(os和dfs)与低危组患者不同。与低风险组的gc患者相比,高风险组gc患者的os和dfs明显较差(图1c和d)。tcga队列中预测1年、3年和5年os的auc分别为0.61、0.64和0.76;tcga队列中预测1年、3年和5年dfs的auc分别为0.61、0.68和0.65(图1e)。接下来,我们调查了tcga队列中args预后评分高或低的患者的临床特征。高args预后评分与肿瘤分期(t期)和肿瘤疾病分期显著相关,因为高危组中出现了更多t4和/或iv期疾病患者(图1f)。
58.2.2 args独立预测os和dfs
59.为了验证新构建的arg信号是否可以作为一个独立的危险因素用于预测gc患者的预后,在高风险评分组和低风险评分组之间测试了各种临床病理参数。在tcga队列中,单变量分析显示args(p=0.002)、t分期(p=0.001)、n分期(p《0.001)、m分期(p=0.004)、肿瘤疾病分期(p《0.001)和肿瘤疾病分级(p=0.021)与os显著相关。多因素cox回归分析证实args、m分期和肿瘤疾病分级是os的独立危险因素。此外,单变量和多变量分析表明,arg作为一个独立的风险因素,也与较差的dfs相关(图2a)。
60.然后根据临床特点将训练组患者分为不同的亚组,并进一步分析ssgsea评分与gc患者os之间的相关性。结果表明,在不同临床特征的分层下,arg特征评分对高分期(iii-iv期)、肿瘤分期(t3-4)和n分期(n1-3)患者的预后有良好影响(图2b)。
61.2.3 tcga gc患者高args和低args预后评分组的基因遗传改变情况
62.为了进一步探讨遗传特征与args预后评分之间的相关性,对基因组突变谱、拷贝数变异(cnv)、甲基化水平进行了分析。如图8所示,分析了36个arg的基因组改变。结果显示,在tcga-gc队列中,vcan(8%)是最常见的arg(图8a左面板)。然后,前20位突变基因出现在高args和低args预后评分组(图8a中幅和右图)。然后选择前十位改变的基因来评估有或没有基因组改变的样本中args风险评分的差异。与ttn和muc16改变组相比,args风险评分显著高于野生型ttn或muc16样本(图8d)。其他突变基因,包括tp53(p=0.094)、lrp1b、arid1a和syne1,没有观察到显著差异。如图8b所示,通过spearman分析,args表达水平与cnv呈正相关。此外,在高args和低args预后评分组中,分析了具有相关系数的前九名args的cnv。高args预后评分组vcan的cnv显著高于低args预后评分组(p《0.01)。而高args预后评分组ptk2的cnv显著低于低args评分组(p《0.05)(图8e)。如图8c所示,通过spearman分析,args相关基因的表达水平与甲基化水平呈负相关(图8c)。分析高精氨酸预后评分组和低精氨酸预后评分组前21位精氨酸的甲基化水平。与高args预后评分组相比,低args预后评分组kcnj8、s100a4、ccnd2、thbd、fstl1、fgfr1和lpl的甲基化水平显著升高,而vegfa、
vav2、tnfrsf21、cxcl6、vtn和pdgfa的甲基化水平显著降低(图8f)。
63.2.4 arg特征评分高的gc患者肿瘤浸润淋巴细胞谱
64.为了进一步研究arg特征风险评分与免疫状态之间的相关性,应用ssgsea计算不同免疫细胞含量的富集程度。如图3a所示,通过spearman的分析,cyt、estimate、hla、icg、til和arg评分之间存在正相关。我们进一步分析了肿瘤纯度与arg评分之间的相关性,发现肿瘤纯度与arg评分之间存在显著的负相关(图3b)。正如我们预期的那样,通过epic对tcga队列中的肿瘤浸润淋巴细胞进行分析,发现高危和低危arg组之间存在统计学差异,包括caf、t细胞cd4+、t细胞cd8+、巨噬细胞、内皮细胞、nk细胞(图3c)。xcell也发现高arg和低arg评分组之间的cafs含量显著差异一致(图3d)。
65.2.5具有高arg特征评分的gc患者的肿瘤浸润淋巴细胞谱
66.为了探索arg特征风险评分与免疫状态之间的相关性,我们使用ssgsea量化不同免疫细胞含量的富集评分。如图3a所示,通过spearman分析观察到cyt、estimate、hla、icg、til和arg评分的正相关性。我们进一步分析了肿瘤纯度与arg评分相关,肿瘤纯度与arg评分呈显著负相关(图3b)如我们所料,通过epic对tcga队列中高风险组和低风险arg组的肿瘤浸润淋巴细胞谱进行分析,显示出统计学差异,包括癌症相关成纤维细胞(caf)、t细胞cd4+、t细胞cd8+、巨噬细胞、内皮细胞、nk细胞(图3c)。与此相同,xcell分析也发现了arg组和arg组之间cafs含量的显著差异性(图3d)。
67.2.6 gc患者cafs亚型与arg信号之间的关联
68.caf是癌间质中最丰富的细胞类型之一,在具有不同arg特征评分的gc患者中进行了分析。根据图4a-h所示的结果,只有基质caf(mcaf)亚型与较差的os和dfs显著相关,而其他亚型,包括血管caf(vcaf)和发育性caf(dcaf)对gc患者的生存率没有显著影响。在三个验证集(gse26253、gse66229和gse26901,见图9a-d)中观察到类似的结果。然后,我们通过spearman评估了arg与各种caf亚型之间的相关性,这表明arg与各种caf(包括mcaf、vcaf和dcafs)呈正相关(图4i-l,图9d)。同时,我们发现arg评分和mcaf含量低的组(arg-mcaf-)的总体存活率最好;相反,arg评分和mcaf含量高的组的患者预后最差(args+mcaf+,图4m&n)。
69.2.7使用外部数据集验证预后arg特征
70.为了进一步评估从训练数据集中识别的arg特征的稳健性和稳定性,我们使用gse26253、gse26091和gse66229数据集进行了生存分析和tme分析。使用相同的公式计算验证队列中患者的风险分数,根据每个队列中风险分数的中值将患者分为低亚组和高亚组。如我们的验证结果所示,与所有三个验证队列中的高危患者相比,低风险亚组中的gc患者的os和dfs显著更好(gse26253:os,p=0.012;gse26901:os,p=0.0012和dfs,p=0.0026;gse66229:os,p=0.00011和dfs,p=0.003)(图5)。此外,在gse26253的高args预后评分组中,基质评分、免疫评分和估计评分显著高于对照组(p《0.001,图10a)。ssgsea评分与tilss、icgs(ctla4、tim3、lag3和ido1)之间的相关性由spearman analysis分析(图10b)进行分析,并且仅在tim3和arg特征评分之间确定了显著相关性(r=0.31,p《0.001)。在gse26901和gse66229队列中观察到类似结果(图11和图12)。
71.2.8基于arg表达谱的聚类分析
72.进行一致性聚类分析,以进一步评估arg在训练队列(tcga)gc患者中的预后意义。训练组的414名gc患者被分为两个亚组,即第1组和第2组。pca进一步验证了簇1和簇2之间
基因表达水平的差异(图6a)。同时,为了研究聚类分析和ssgesa评分的这些分类方法之间的重叠,我们使用维恩图来显示结果(图6b)。生存分析显示,与第一组患者相比,第二组患者的os显著更好(图6c和d)。同时,第2组的基质评分、免疫评分和评估评分以及tim3水平显著降低(图6f)。此外,集群1的b细胞、caf、cd4t细胞、内皮细胞、巨噬细胞和非特征化细胞的含量显著较高(图6g),xcell分析进一步验证了这一点(图6h)。
73.2.9args相关预后预测因子的建立和验证
74.首先,在训练集中,根据deg的表达水平,使用四种机器学习算法识别最关键的子类型相关特征。lasso、rf、svm和xgboost分析分别用于识别33、114、80和110个基因。维恩图确定了十个关键基因,这四个特征选择算法共享了这些基因(图6i)。然后,通过基于多变量套索的logistic回归分析构建诊断预测模型。
75.arg亚型预测模型的计算公式如下:
76.预测分值=5.869+0.852
×
(dclk1的表达水平)+0.295
×
(ptgis的表达水平)+0.340
×
(nudt10的表达水平)+0.598
×
(zfhx4的表达水平)+0.290
×
(pcdh9的表达水平)+0.211
×
(chrdl1的表达水平)+0.073
×
(nlgn1的表达水平)+0.298
×
(agtr1的表达水平)+0.221
×
(cntn1表达水平)+0.261
×
(ecrg4表达水平)。
77.roc分析显示,在训练集中分离i型和ii型亚型的auc为0.994(图6j)。此外,亚型预测因子在区分测试集中评估的亚型方面也表现出色,auc为0.979(图6k)。k-m生存分析显示,在gse26901队列中,具有高危评分的gc患者的os和pfs明显较差(图6l-m)。gse26901队列中预测1年、3年和5年os的auc分别为0.7、0.65和0.64,gse26901队列中预测1年、3年和5年dfs的auc分别为0.66、0.62和0.59(图6n-o)。在gse66229队列中,风险评分与患者生存率之间的关联也被确定为高风险评分患者的dfs和os显著较差(图6p-o)。在gse66229队列中,构建的arg信号的预测效果被证明是稳定且良好的(预测1年、3年和5年os的auc分别为0.59、0.63和0.64;预测1年、3年和5年dfs的auc分别为0.57、0.64和0.65,图6r-s)。
78.2.10免疫治疗和靶向治疗的分子特征和疗效鉴定
79.如图7所示,gse26901中gc患者的复发率存在显著差异,且疾病复发患者的风险评分显著较高(图7a)。同时,在gse66229中,gc的emt亚型显著高于其他三种gc类型,包括msi、tp53阳性和tp53阴性(图7b)。与cin、pole、msi和ebv感染亚型相比,gs亚型的预测能力明显更高(图7c)。与高args评分组相比,低args评分组表现出更广泛的肿瘤突变负担(图7d-e)。
80.而在膀胱癌imvigor210数据集中kaplan
–
meier曲线显示,低arg亚型评分组患者的预后比高分组患者好得多(图7f)。
81.与高args评分组相比,低args评分组表现出更广泛的肿瘤突变负荷(图7d-e)。而肿瘤突变负荷是与免疫治疗效果直接相关的。本发明以膀胱癌为例,研究args评分与免疫治疗的关系。膀胱癌imvigor210数据集中,对抗pd-1/l1抗体免疫治疗有反应的患者的arg评分低于无反应的患者(p=0.051,图7g)。此外,突变计数、fgfr4、her2与arg亚型预测得分呈显著负相关(图7h)。
82.值得注意的是,与血管生成特征一致的是,arg亚型评分高的患者的一些vegf和pdgf受体抑制剂的ic50水平显著降低,这些抑制剂可以抑制血管生成和肿瘤的发展,包括舒尼替尼、索拉非尼、帕佐帕尼和阿西替尼。尽管所有这些靶向治疗方案尚未在胃癌中获得批准,但这表明具有高风险特征的gc患者可能从抗血管生成治疗中获得更大的益处(图
7i)。
83.3.结论
84.本发明首先系统地评估了36args mrna的表达与tcgagc样本中存活率(os和dfs)之间的相关性。tcga gc患者的生存概率与ssgsea评分呈负相关。通过spearman分析,tcga-gc患者的基质和免疫检查点基因(icg)表达与ssgsea评分呈正相关。此外,本发明发现癌相关成纤维细胞和内皮细胞的浸润强度与ssgsea评分呈正相关。以往的研究表明,肿瘤相关的成纤维细胞和内皮细胞在肿瘤细胞增殖、肿瘤血管生成、逃避免疫监视和转移中起着重要作用。此外,caf与血管生成显著相关。在高和低cafs密度组中观察到gc患者生存率的显著差异。此外,本发明发现,与低精氨酸/低mcafs亚组相比,高精氨酸/高mcafs亚组的gc患者生存率显著降低。三个验证队列(gse26253、gse26901和gse66229)用于验证tcga数据库的结果。此外,lasso、svm、rfb和xgboostmachine学习方法被用于识别和定义10个最关键的亚型相关基因作为亚型预测因子,并进一步促进临床实践中区分这两个亚型。本发明研究结果为预测gc患者的预后和生存提供了一种手段,并可能为gc患者的诊断和免疫治疗提供有希望的靶点。
85.血管生成在包括生殖、胚胎发育和伤口愈合在内的生理和病理过程的组织修复和再生中起着至关重要的作用,更重要的是,它是癌症发展的标志性事件之一。在正常生理条件下,内皮细胞对血管生成信号敏感,并通过维持高度可塑性参与血管生成。相反,在包括癌症、类风湿性关节炎和动脉粥样硬化在内的许多疾病状态中,异常血管生成将进一步加速这些疾病的恶化,并被视为这些疾病状态的标志。研究表明,肿瘤血管生成对肿瘤细胞的持续生存和发展至关重要,对肿瘤细胞的生长、侵袭和转移至关重要。本发明的结果表明,与低args评分组相比,高args评分组gc患者的os和dfs显著降低。这些结果进一步证明,args信号是gc患者os和dfs的重要预后和预测标志物。
86.以往的研究表明,基因表达的改变与癌症的预后相关。本发明中,我们的结果显示args表达与cnv、甲基化水平和突变没有显著相关性。这些结果表明,cnv、甲基化水平和args突变对提高预测准确性没有太大影响。肿瘤微环境与肿瘤发生密切相关,因为它包含肿瘤细胞,可通过循环和淋巴系统与周围细胞相互作用,进一步干扰癌症的发展和进展。tme主要由三种细胞成分组成,包括内皮细胞、免疫细胞(粒细胞、淋巴细胞和巨噬细胞)和成纤维细胞。来自附近内源性组织基质的细胞被癌细胞吸收,在肿瘤形成的关键步骤中发挥重要作用。癌相关成纤维细胞(caf)是肿瘤基质的主要组成部分,在促进癌细胞和tme之间的串扰方面至关重要。
87.在本发明中,早期结果表明,arg的特征变化对提高预测精度没有太大影响。有趣的是,本发明的结果显示基质评分与ssgesa评分显著正相关。研究表明,caf与癌症和肿瘤发生的不良预后显著相关。本发明结果表明,与低args评分相比,高args评分的caf数量显著增加。与低密度mcaf相比,高密度mcaf患者的os和dfs显著降低。此外,本发明发现与高cafs相关的高arg导致gc患者os降低。这些结果表明cafs可能是gc预后的生物标志物。
88.总之,本发明的研究证实了caf与gc血管生成过程相关基因表达的相关性。此外,在本发明中,使用gc将患者分为ⅰ型亚型和ⅱ型亚型组,并预测不同亚型患者对免疫治疗的反应。这些结果为未来选择免疫治疗患者提供了一种潜在的方法。它们可以作为预测肿瘤患者预后和诊断的潜在生物标志物发挥重要作用,并为肿瘤患者的靶向治疗提供新的视
角。
89.以上已对本发明创造的较佳实施例进行了具体说明,但本发明创造并不限于所述实施例,熟悉本领域的技术人员在不违背本发明创造精神的前提下还可做出种种的等同的变型或替换,这些等同的变型或替换均包含在本技术权利要求所限定的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1