一种注射用左奥硝唑衍生物乳状制剂及其制备方法与流程
1.本发明属于药物制剂领域,具体涉及一种左奥硝唑衍生物乳状制剂及其制备方法。
背景技术:
2.硝基咪唑类抗菌药物是一类人工合成的抗菌药物,对厌氧菌及原虫有独特的杀灭作用,与其他抗生素联合应用于临床的各个领域。自20世纪50年代第一代硝基咪唑类药物甲硝唑问世后,相继上市的第二代替硝唑和第三代奥硝唑,均在临床上广泛用于由厌氧菌、阿米巴原虫、贾滴虫、毛滴虫等引起的各种感染。
3.目前临床上使用的第三代硝基咪唑类药物奥硝唑主要为消旋体,含有等量的左旋体和右旋体,注射液为主要剂型。发明专利cn1332662c通过对奥硝唑的左旋体进行药代动力学、药效学、毒理学、一般药理学等试验研究,发现左旋奥硝唑药代动力学特性优于右旋奥硝唑和消旋奥硝唑,且中枢毒性低于右旋奥硝唑和消旋奥硝唑,将左旋奥硝唑用于制备抗厌氧菌感染药物更具有实用性。
4.上市产品左奥硝唑氯化钠注射液作用于厌氧菌、阿米巴原虫、贾弟鞭毛虫和毛滴虫细胞的dna,通过其分子的硝基在无氧环境中还原成氨基,或通过自由基的形成,使dna螺旋结构断裂,阻断其转录复制而衰亡,从而达到抗菌抗感染的目的。与替硝唑、甲硝唑等硝基咪唑类药物相比,左奥硝唑抗感染优势更为明显,且副作用更小。临床试验已证明,左奥硝唑在抗厌氧菌感染临床疗效与奥硝唑相当的基础上,临床总不良反应发生率显著降低,仅为奥硝唑的1/15,尤其在神经安全性方面具有显著优势。
5.在多年的临床使用中,左奥硝唑氯化钠注射液虽然抗厌氧菌疗效显著、神经毒性较消旋体注射液有所减轻,但仍存在其他尚未解决的问题。
6.由于左奥硝唑水溶性极差,制成输液制剂的ph约为3.5,酸度值偏高,超过人体耐受ph 4.0~9.0,注射期间强烈的刺激性可干扰血管内膜的正常代谢和机能,对血管内皮细胞造成损伤、诱发血小板聚集,继发血栓性链式反应,轻则产生刺痛感,重则发生不同程度的静脉炎症,如部分患者伴有红肿、静脉条索状改变,导致患者依从性较差,尤其血管壁硬化或较薄的老人及儿童。
7.在临床使用中,左奥硝唑氯化钠注射液仅可采用输注的给药方式,且为适度缓解给药部位刺激性,输注时长须控制在0.5~1h间,不可采用静脉推注,患者顺应性不佳的同时,消耗更多的医疗资源,其临床应用受限。
8.因此,对于硝基咪唑类抗厌氧菌注射剂,临床需要更加安全、更加便利、能够增加患者顺应性的产品。
技术实现要素:
9.本发明的目的在于提供一种注射用左奥硝唑衍生物乳状制剂及其制备方法。从产品剂型及化合物结构两个方面协同解决现存临床问题。
10.注射液的给药部位刺激性来源主要为:溶剂主要成分、溶剂ph、化合物自身性质。基于市售产品质量情况,本发明制备ph在4.0~9.0、甚至接近中性的注射液,消除ph不适宜因素;进一步地,使用乳剂剂型将活性成分包裹在油相内部,适当延迟释药时间,缓解化合物带来的刺激性,更进一步,根据剂型对化合物的溶解度要求,将左奥硝唑的结构进行修饰,制备成具有一定代谢速率的前药,彻底避免左奥硝唑在给药部位释放。
11.本发明主要是通过如下技术方案实现的:
12.本发明提供一种含左奥硝唑衍生物的乳状制剂,其包含:
13.(a)结构式(1)所示的化合物,其消旋体、立体异构体、药学上可接受的盐或溶剂合物,或者其药学上可接受的盐的溶剂合物:
[0014][0015]
(b)乳化剂;
[0016]
(c)注射用油。
[0017]
根据本发明实施方案,以乳状制剂总体积为100%计,活性成分的质量百分比为0.1-10%(w/v),优选为1-5%(w/v)。
[0018]
适合用于本发明的乳化剂选自天然乳化剂,选自大豆卵磷脂、蛋黄卵磷脂、氢化卵磷脂、饱和和不饱和c
12-18
脂肪酰磷脂酰胆碱中的一种、两种或更多种的组合;
[0019]
根据本发明实施方案,所述乳化剂选自天然蛋黄卵磷脂或/和天然大豆卵磷脂,质量百分比为0.5-5%(w/v),优选为1-2%(w/v)。
[0020]
适合用于本发明的油为可注射用油酯,选自大豆油、红花油、棉籽油、橄榄油、芝麻油、椰子油、蓖麻油、沙棘油、月见草油、玉米油、鸦胆子油、紫苏油、葡萄籽油、茶油、棕榈油、花生油、中链油(中链甘油三酯)、长链甘油三酯、油酸乙酯、乙酰化单甘油酯、丙二醇双酯、亚油酸甘油酯或聚乙二醇月桂酸甘油酯,或其中两种或更多种的组合。
[0021]
根据本发明实施方案,所述油选自中链甘油三酸酯、大豆油、花生油的一种或几种,质量百分比为5-30%(w/v),优选10-20%(w/v)。
[0022]
根据本发明实施方案,所述乳状制剂还包括稳定剂,所述稳定剂可选自油酸、油酸钠,kolliphor hs15、聚山梨酯、多库酯钠、去氧胆酸、泊洛沙姆中的一种或几种。
[0023]
根据本发明实施方案,所述乳状制剂还包括等渗调节剂,所述等渗调节剂选自蔗糖、葡萄糖、山梨醇、木糖醇、氯化钠、甘油中的一种或几种。
[0024]
根据本发明实施方案,所述乳状制剂还可以包括ph调节剂,所述ph调节剂选自枸橼酸、盐酸、柠檬酸、富马酸、赖氨酸、酒石酸、组氨酸枸橼酸钠、氢氧化钠、柠檬酸钠、磷酸二氢钠、磷酸氢二钠中的一种或几种。
[0025]
本发明还提供一种含左奥硝唑衍生物的乳状制剂的制备方法,包括以下步骤:
[0026]
(1)油相制备:向注射用油中加入乳化剂、脂溶性稳定剂,高速剪切使其溶解,再加入左奥硝唑衍生物,搅拌溶解混匀,即得;
[0027]
(2)水相制备:向注射用水中加入等渗调节剂、水溶性稳定剂,必要时加入ph调节剂,搅拌溶解,即得;
[0028]
(3)初乳制备:将步骤(2)水相加入步骤(1)油相中,于水浴保温下高速剪切分散,形成初乳;
[0029]
(4)终乳制备:对步骤(3)所得初乳进行高压均质,得终乳,必要时加入ph调节剂调节终乳ph;
[0030]
(5)过滤、灌装、充氮、湿热灭菌,即得。
[0031]
本发明制备的乳状制剂适宜于肠胃外给药方式给药,肠胃外给药包括静脉内、动脉内、皮下、腹膜内或肌内注射或输注;或颅内例如鞘内或脑室内给药。可按单次大剂量形式肠胃外给药,或可通过例如连续灌注泵给药。或颅内例如鞘内或脑室内给药;注射剂常用容器有玻璃安瓿、西林瓶、塑料安瓿、预灌封注射器等。
[0032]
有益效果:
[0033]
本发明利用未经报道过的新化合物左奥硝唑衍生物为活性成分,通过研究活性成分的理化性质,综合考虑其体内外代谢试验,进一步研究制剂处方,从而获得了一种安全、稳定的含左奥硝唑衍生物的乳状制剂,采用本发明技术方案制备的乳状注射液可避免现有产品对血管的刺激性、减少静脉炎的发生,高载药量可实现静脉推注的给药方式,增加患者的顺应性、节约医疗资源;并可通过控制平均粒径,实现不同的释药速率,满足多样化的临床治疗方案。
附图说明
[0034]
图1为左奥硝唑衍生物的核磁氢谱图。
[0035]
图2为左奥硝唑衍生物有关物质测定高效液相色谱图。
[0036]
图3为本发明左奥硝唑衍生物乳状制剂的典型粒度分布图。
[0037]
图4为本发明左奥硝唑衍生物乳状制剂的典型zeta电位测定结果图。
[0038]
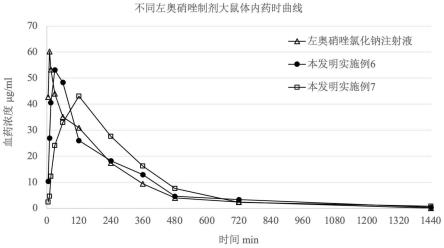
图5为左奥硝唑氯化钠注射液和左奥硝唑衍生物乳状注射液的血药浓度-时间曲线。
具体实施方式
[0039]
下面结合具体实施例对本发明作进一步详述,以下实施例只是描述性的,不是限定性的,不能以此限定本发明的保护范围,如无特别说明,所用原料均可通过市售或自制获得。
[0040]
本发明中涉及的主要检测方法:
[0041]
一、ph测定:
[0042]
照ph值测定法(中国药典2020版通则0631)测定。
[0043]
二、有关物质测定
[0044]
取样品适量用70%乙腈水溶液稀释至左奥硝唑衍生物浓度约为0.5mg/ml,作为供试品溶液。照高效液相色谱法(中国药典2020版通则0512)测定。采用agilent zorbax c18
(250*4.6mm,5μm)色谱柱,以水为流动相a,以乙腈为流动相b,按下表进行梯度洗脱:
[0045][0046][0047]
流速:1.0ml/min;检测波长:318nm;柱温:35℃;进样量:10μl,记录色谱图,分析杂质情况。
[0048]
典型有关物质图谱见图2。
[0049]
三、粒度、zeta电位测定:
[0050]
取本品乳状注射液约50μl稀释于4ml经过滤的纯化水中,振摇使其混合均匀,得供试品混合物。照粒度和粒度分布测定法(中国药典2020版通则0982第三法),采用基于瑞利散射理论的动态光散射光学粒度分析仪pss nicomp z3000,采用下列参数:光学强度300khz;折射角90
°
;时间5分钟,进行粒度和zeta电位检测。记录粒径的高斯分布图,并对平均粒径、pi、zeta电位等数据进行分析。典型粒径图谱见图3,zeta电位测定结果见图4。
[0051]
试验例1左奥硝唑衍生物的合成:
[0052][0053]
中间体1的合成:称取左奥硝唑(5.036g,22.93mmol)加入瓶中,加入dcm 20ml室温搅拌溶解,再称取py(3.609g,45.62mmol),dmap(2.811g,23.01mmol)依次加入反应瓶中,最后称取乙酸酐(3.514g,34.42mmol)缓慢滴入反应瓶中,保持搅拌1h,tlc监控至原料消失。后处理,加入50ml水和30ml dcm萃取分液取有机层,有机层再分别用50ml饱和nahco3与饱和食盐水洗,无水na2so4干燥,然后减压浓缩得白色固体(4.367g,16.69mmol),收率为72.8%。
[0054]
结构式(1)化合物的合成:称取中间体1(1.056g,4.04mmol)加入瓶中,加入dcm6ml室温搅拌溶解,反应瓶用ar气置换3次,将反应瓶移至冷阱中-78℃搅拌,将(1.162g,8.17mmol)二异丁基氢化铝缓慢滴入反应瓶中,滴毕保持搅拌1h,称取py(0.965g,12.20mmol),dmap(1.004g,8.22mmol)分别用2ml dcm稀释后缓慢滴入反应瓶中,滴毕再称取乙酸酐(2.496g,24.45mmol)滴入反应瓶中,保持搅拌20h,开始后处理,加入dcm 40ml、饱和nh4cl 50ml萃取分液取有机层,有机层再分别用50ml水与饱和食盐水洗,无水na2so4干燥,旋蒸得油状物,再硅胶柱层析分离,使用正己烷:乙酸乙酯4:1的混合溶剂冲柱,旋蒸浓
缩,最终得产物(0.272g,0.89mmol),收率为22.0%。1hnmr(400mhz,cdcl3)δ7.97(d,1h,j=11.3hz),δ5.82-5.44(m,1h),δ4.73-4.60(m,1h),δ4.38-4.09(m,2h),δ3.76-3.64(m,2h),δ2.53(s,3h),δ1.90(d,3h,j=95.0hz),δ1.25(dd,3h,j=5.3hz,j=69.2hz);其核磁氢谱图如图1所示。
[0055]
ms(esi-ms)calcd for c
11h16
cln3o5[m+h]+306.08,found 306.0
[0056]
试验例2:化合物稳定性研究
[0057]
实验方法:取本发明结构式(1)化合物适量置于西林瓶中,分别在高温(60℃,敞口)、光照(4500lx,25℃,敞口)和高湿(75%rh和92.5%rh,敞口)条件下放置,分别于0、1、5、8天取样,考察化合物纯度及有关物质变化情况,具体结果如下表所示:
[0058][0059]
由试验结果可知,化合物在高温(60℃,敞口)、光照(4500lx,25℃,敞口)和高湿(75%rh和92.5%rh,敞口)条件下放置8天后,光照和高温条件下杂质略有增加,但含量都在98%以上,高湿条件下杂质与留样前一致,表明该化合物在上述条件下8天内稳定性良好,可支持将其制成乳状注射液的相关研究。
[0060]
试验例3:油相筛选
[0061]
药物制剂领域常用的油类辅料主要有花生油、中链甘油三酸酯、稻米油、蓖麻油、葵花仁油、芝麻油、茶油、玉米胚芽油、大豆油等,本发明中,选择芝麻油、中链甘油三酸酯、大豆油、葵花仁油和花生油,考察化合物在上述油中的溶解情况,选出优选的油相。
[0062]
实验方法:
[0063]
称取化合物约0.1g,加入约1g的上述油中,室温搅拌,观察溶解情况。若溶清,则继续加入0.1g化合物,直至不能溶解至澄清。
[0064]
油种类溶解情况芝麻油加至0.3g化合物开始浑浊中链甘油三酸酯加至0.6g化合物开始浑浊大豆油加至0.4g化合物开始浑浊
蓖麻油加至0.2g化合物开始浑浊花生油加至0.3g化合物开始浑浊
[0065]
上述试验结果表明:化合物在中链甘油三酸酯中的溶解度要明显大于在芝麻油、大豆油、蓖麻油、花生油中的溶解度,结合目前广泛应用于临床的载药乳状注射液多采用大豆油或中链甘油三酸酯作为油相,后续实施例仅考察以中链甘油三酸酯作为油相对乳状注射液稳定性等的影响。
[0066]
下面结合具体实施例对本发明进行进一步说明。
[0067]
实施例1
[0068]
处方组成:
[0069][0070][0071]
制备方法:
[0072]
(1)油相制备:60℃水浴保温下,向中链甘油三酸酯中加入蛋黄卵磷脂lipod e80、油酸,高速剪切使其溶解,再加入左奥硝唑衍生物即结构式(1)所示化合物,搅拌溶解混匀,即得;
[0073]
(2)水相制备:60℃水浴保温下,向注射用水中加入甘油,搅拌溶解,即得;
[0074]
(3)初乳制备:将步骤(2)水相加入步骤(1)油相中,于60℃水浴保温下高速剪切分散1min,形成初乳;
[0075]
(4)终乳制备:对步骤(3)所得初乳进行高压均质,800-1000bar压力下均质90s,得终乳,加入1m naoh溶液调节ph至7.37;
[0076]
(5)过滤、灌装至西林瓶、充氮、加塞轧盖、116℃灭菌40min,即得。
[0077]
实施例2
[0078]
处方组成:
[0079]
原辅料投料量g左奥硝唑衍生物0.7中链甘油三酸酯10蛋黄卵磷脂pl-100m1.2油酸0.03甘油2.2注射用水85.87
[0080]
制备方法:
[0081]
(1)油相制备:60℃水浴保温下,向中链甘油三酸酯中加入蛋黄卵磷脂pl-100m、油
酸,高速剪切使其溶解,再加入左奥硝唑衍生物即结构式(1)化合物,搅拌溶解混匀,即得;
[0082]
(2)水相制备:60℃水浴保温下,向注射用水中加入甘油,搅拌溶解,即得;
[0083]
(3)初乳制备:将步骤(2)水相加入步骤(1)油相中,于60℃水浴保温下高速剪切分散1min,形成初乳;
[0084]
(4)终乳制备:对步骤(3)所得初乳进行高压均质,800-1000bar压力下均质90s,得终乳,加入1m naoh溶液调节ph至7.46;
[0085]
(5)过滤、灌装至西林瓶、充氮、加塞轧盖、116℃灭菌40min,即得。
[0086]
实施例3
[0087]
处方组成:
[0088]
原辅料投料量g结构式(1)化合物1.4中链甘油三酸酯10蛋黄卵磷脂pl-100m1.2油酸0.03甘油2.2注射用水85.17
[0089]
制备方法:
[0090]
(1)油相制备:60℃水浴保温下,向中链甘油三酸酯中加入蛋黄卵磷脂pl-100m、油酸,高速剪切使其溶解,再加入左奥硝唑衍生物即结构式(1)化合物,搅拌溶解混匀,即得;
[0091]
(2)水相制备:60℃水浴保温下,向注射用水中加入甘油,搅拌溶解,即得;
[0092]
(3)初乳制备:将步骤(2)水相加入步骤(1)油相中,于60℃水浴保温下高速剪切分散1min,形成初乳;
[0093]
(4)终乳制备:对步骤(3)所得初乳进行高压均质,800-1000bar压力下均质90s,得终乳,加入1m naoh溶液调节ph至7.68;
[0094]
(5)过滤、灌装至西林瓶、充氮、加塞轧盖、116℃灭菌40min,即得。
[0095]
实施例4
[0096]
处方组成:
[0097]
原辅料投料量g结构式(1)化合物1.4中链甘油三酸酯10蛋黄卵磷脂pl-100m2油酸0.03甘油2.2注射用水84.37
[0098]
制备方法:
[0099]
(1)油相制备:60℃水浴保温下,向中链甘油三酸酯中加入油酸,高速剪切使其溶解,再加入左奥硝唑衍生物即结构式(1)化合物,搅拌溶解混匀,即得;
[0100]
(2)水相制备:60℃水浴保温下,向注射用水中加入甘油,搅拌溶解,再加入蛋黄卵磷脂pl-100m,搅拌至目视分散均匀,即得;
[0101]
(3)初乳制备:将步骤(2)水相加入步骤(1)油相中,于60℃水浴保温下高速剪切分散1min,形成初乳;
[0102]
(4)终乳制备:对步骤(3)所得初乳进行高压均质,800-1000bar压力下均质90s,得终乳,加入1m naoh溶液调节ph至7.62;
[0103]
(5)过滤、灌装至西林瓶、充氮、加塞轧盖、116℃灭菌40min,即得。
[0104]
实施例5
[0105]
处方组成:
[0106]
原辅料投料量g左奥硝唑衍生物5中链甘油三酸酯15蛋黄卵磷脂pl-100m2油酸钠0.5甘油2.3注射用水75.20
[0107]
制备方法:
[0108]
(1)油相制备:60℃水浴保温下,向中链甘油三酸酯中加入蛋黄卵磷脂pl-100m,高速剪切使其溶解,再加入左奥硝唑衍生物即结构式(1)化合物,搅拌溶解混匀,即得;
[0109]
(2)水相制备:60℃水浴保温下,向注射用水中加入甘油、油酸钠,搅拌溶解,即得;
[0110]
(3)初乳制备:将步骤(2)水相加入步骤(1)油相中,于60℃水浴保温下高速剪切分散1min,形成初乳;
[0111]
(4)终乳制备:对步骤(3)所得初乳进行高压均质,800-1000bar压力下均质120s,得终乳,加入1m hcl溶液调节ph至6.98;
[0112]
(5)过滤、灌装至西林瓶、充氮、加塞轧盖、116℃灭菌40min,即得。
[0113]
实施例6
[0114]
处方组成:
[0115]
原辅料投料量g左奥硝唑衍生物7中链甘油三酸酯18蛋黄卵磷脂pl-100m3油酸钠0.5甘油2.2注射用水69.30
[0116]
制备方法:
[0117]
(1)油相制备:60℃水浴保温下,向中链甘油三酸酯中加入蛋黄卵磷脂pl-100m,高速剪切使其溶解,再加入左奥硝唑衍生物即结构式(1)化合物,搅拌溶解混匀,即得;
[0118]
(2)水相制备:60℃水浴保温下,向注射用水中加入甘油、油酸钠,搅拌溶解,即得;
[0119]
(3)初乳制备:将步骤(2)水相加入步骤(1)油相中,于60℃水浴保温下高速剪切分散1min,形成初乳;
[0120]
(4)终乳制备:对步骤(3)所得初乳进行高压均质,800-1000bar压力下均质120s,
得终乳,加入1m hcl溶液调节ph至7.19;
[0121]
(5)过滤、灌装至西林瓶、充氮、加塞轧盖、116℃灭菌40min,即得。
[0122]
实施例7
[0123]
处方组成:
[0124]
原辅料投料量g左奥硝唑衍生物7中链甘油三酸酯18蛋黄卵磷脂pl-100m1.2油酸钠0.5泊洛沙姆1880.5甘油2.2注射用水70.60
[0125]
制备方法:
[0126]
(1)油相制备:60℃水浴保温下,向中链甘油三酸酯中加入蛋黄卵磷脂pl-100m,高速剪切使其溶解,再加入左奥硝唑衍生物即结构式(1)化合物,搅拌溶解混匀,即得;
[0127]
(2)水相制备:60℃水浴保温下,向注射用水中加入甘油、油酸钠、泊洛沙姆188,搅拌溶解,即得;
[0128]
(3)初乳制备:将步骤(2)水相加入步骤(1)油相中,于60℃水浴保温下高速剪切分散1min,形成初乳;
[0129]
(4)终乳制备:对步骤(3)所得初乳进行高压均质,800-1000bar压力下均质90s,得终乳,加入1m hcl溶液调节ph至7.19;
[0130]
(5)过滤、灌装至西林瓶、充氮、加塞轧盖、116℃灭菌40min,即得。
[0131]
实施例8
[0132]
处方组成:
[0133]
原辅料投料量g左奥硝唑衍生物10中链甘油三酸酯30蛋黄卵磷脂pl-100m5聚山梨酯800.5甘油2.2注射用水52.30
[0134]
制备方法:
[0135]
(1)油相制备:60℃水浴保温下,向中链甘油三酸酯中加入蛋黄卵磷脂pl-100m,高速剪切使其溶解,再加入左奥硝唑衍生物即结构式(1)化合物,搅拌溶解混匀,即得;
[0136]
(2)水相制备:60℃水浴保温下,向注射用水中加入甘油、聚山梨酯80,搅拌溶解,即得;
[0137]
(3)初乳制备:将步骤(2)水相加入步骤(1)油相中,于60℃水浴保温下高速剪切分散1min,形成初乳;
[0138]
(4)终乳制备:对步骤(3)所得初乳进行高压均质,800-1000bar压力下均质240s,
得终乳,加入1m hcl溶液调节ph至8.17;
[0139]
(5)过滤、灌装至西林瓶、充氮、加塞轧盖、116℃灭菌40min,即得。
[0140]
左奥硝唑氯化钠注射液规格0.5g:100ml,为仅能静脉滴注的大容量注射剂,根据上述实施例处方组成,本发明能够达到以左奥硝唑计最高约0.5g:7ml的载药量,完全满足静脉推注的药液体积要求。
[0141]
对比例1
[0142]
处方组成:
[0143]
原辅料投料量g左奥硝唑衍生物15中链甘油三酸酯30蛋黄卵磷脂pl-100m5油酸0.1聚山梨酯801甘油2.2注射用水46.70
[0144]
制备方法:
[0145]
(1)油相制备:60℃水浴保温下,向中链甘油三酸酯中加入蛋黄卵磷脂pl-100m、油酸,高速剪切使其溶解,再加入左奥硝唑衍生物即结构式(1)化合物,搅拌溶解混匀,即得;
[0146]
(2)水相制备:60℃水浴保温下,向注射用水中加入甘油、聚山梨酯80,搅拌溶解,即得;
[0147]
(3)初乳制备:将步骤(2)水相加入步骤(1)油相中,于60℃水浴保温下高速剪切分散1min,形成初乳;
[0148]
(4)终乳制备:对步骤(3)所得初乳进行高压均质,800-1000bar压力下均质240s,得终乳,加入1m hcl溶液调节ph至7.51;
[0149]
(5)过滤、灌装至西林瓶、充氮、加塞轧盖、116℃灭菌40min,即得。
[0150]
对比例2
[0151]
处方组成:
[0152][0153][0154]
制备方法:
[0155]
(1)油相制备:60℃水浴保温下,向中链甘油三酸酯中加入蛋黄卵磷脂pl-100m,高速剪切使其溶解,再加入左奥硝唑衍生物即结构式(1)化合物,搅拌溶解混匀,即得;
[0156]
(2)水相制备:60℃水浴保温下,向注射用水中加入甘油、泊洛沙姆188,搅拌溶解,
即得;
[0157]
(3)初乳制备:将步骤(2)水相加入步骤(1)油相中,于60℃水浴保温下高速剪切分散1min,形成初乳;
[0158]
(4)终乳制备:对步骤(3)所得初乳进行高压均质,800-1000bar压力下均质120s,得终乳,加入1m hcl溶液调节ph至6.82;
[0159]
(5)过滤、灌装至西林瓶、充氮、加塞轧盖、116℃灭菌40min,即得。
[0160]
试验例3:稳定性考察
[0161]
取实施例1~8、对比例1-2样品于4℃和25℃条件下稳定性放样,放置30天后考察粒径和电位的变化情况。具体结果见下表:
[0162][0163]
上述检测粒径及电位样品均为淡黄色乳状液,流动性良好,其中破乳样品为目视瓶壁附着油滴,不再检测。其中,对比例1中左奥硝唑衍生物用量过高,导致制备的乳剂粒径较大,易破乳。而对比例2中乳化剂蛋黄卵磷脂用量较少,起不到良好的乳化作用,导致制剂灭菌后即破乳。
[0164]
试验例4:本发明乳状注射液在大鼠体内药动学研究
[0165]
取本发明实施例6、实施例7乳状注射液和市售左奥硝唑氯化钠注射液对大鼠进行尾静脉注射给药,每组3只大鼠,给药前禁食,剂量为以左奥硝唑计50mg/kg,分别在给药后的5、10、15、30、60、120、240、360、480、720和1440min,通过大鼠眼底静脉取血并置于肝素处理的试管中。将全血8000rpm离心5min,分取血浆样品置于-80℃下保存,分析测定血浆中左奥硝唑的浓度。考察大鼠血浆中左奥硝唑与时间的变化关系,图5为左奥硝唑氯化钠注射液和左奥硝唑衍生物乳状注射液血药浓度-时间曲线。
[0166]
按照设计原理,本发明实施例6、实施例7样品注射后均出现了一定程度的达峰延迟,可抑制左奥硝唑暴露于注射部位。对于实施例6样品,血药浓度达峰时间较左奥硝唑氯化钠注射液稍稍后移,药时曲线下面积与左奥硝唑氯化钠注射液无显著差异,说明其抗菌效果可与其一致。对于实施例7样品,粒径显著增加导致释药速率明显减缓,血药浓度达峰时间约为2小时,且最高血药浓度有所降低,但药时曲线下面积几乎未受影响,这为肝肾功能异常患者提供了更加安全的血药浓度空间;为术前预防提供了足够的用药时间;为本品
与其他静脉注射药物联合使用留出一定空间,避免两者同时达到最高血药浓度增加患者代谢负担。患者顺应性及医师临床用药灵活性均有增加。
[0167]
以上所述仅为本发明的较佳实施例,并非用以限定本发明的实质技术内容范围,本发明的实质技术内容是广义地定义于申请的权利要求范围中,任何他人完成的技术实体或方法,若是与申请的权利要求书范围所定义的完全相同,也或是一种等效的变更,均将被视为涵盖于该权利要求范围之中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1