壳聚糖/普鲁兰多糖微纤维载体及制备方法、缓释药物及制备方法、药物组合物
1.本发明是关于缓释药物载体技术,特别是关于一种壳聚糖/普鲁兰多糖微纤维载体及制备方法、缓释药物及制备方法、药物组合物。
背景技术:
2.药物缓释系统是在1952年由smith kline提出的一种新型给药系统,它以物理吸附或者化学吸附的方法将不同类型的药物与高分子载体结合,然后通过溶解、扩散与溶蚀等形式将药物释放出来,通过对聚合物载体进行设计,使药物在人体内长期缓慢释放,以实现药效发挥的最大化,从而有效地治疗疾病。但是传统的药物输送方式会出现药物浓度波动很大的现象,最开始药物浓度会很高,甚至有可能超过人体能接受的上限,从而使人体产生副作用。开始释放大量药物后,后续的释放速度会逐渐减小,使得体内的药物含量会低于有效剂量,导致药物失去治疗效果。因此,传统的药物输送方式达不到持续释放药物的目的。而少量多次的给药方式虽然能够在一定程度上控制患者体内的药物浓度,但操作过于繁琐。对比之下,再引入药物缓释系统后,药物可以在一定时间内保持恒定的速度从载体中释放出来,从而使人体体液中药物浓度保持在有效范围之内,波动小,从而防止药物浓度的极差大,又能保证患者疾病得到治疗,有效削弱药物的毒副作用;并且药物缓释系统可以根据药物特性设计不同载体,从而实现对药物剂量的有效装载,降低药物对人体的伤害,减少用药的次数,进而缓解不断用药给患者带来的痛苦;还可以根据药物发挥作用的条件来实现药物的缓慢、精准释放,达到药物治疗的目的,延长药物治疗的时长,保障药物治疗的稳定性,从而减少人力、物力及财力的消耗。
3.对乙酰氨基酚(aap)是常见的一类抗炎药物,可用于治感冒发热,关节痛、神经痛、偏头痛等中轻度疼痛,但是使用过量会造成严重的肝损伤,在日常生活中极易被过量使用。因此如何改善其有效浓度的持续时间,是目前的主要研究方向。
4.公开于该背景技术部分的信息仅仅旨在增加对本发明的总体背景的理解,而不应当被视为承认或以任何形式暗示该信息构成已为本领域一般技术人员所公知的现有技术。
技术实现要素:
5.本发明的目的在于提供一种壳聚糖/普鲁兰多糖微纤维载体及其制备方法、缓释药物及制备方法、药物组合物,具有良好的长效性和成型加工性能,有利用长效地实现乙酰氨基酚(aap)类缓释药物的血药峰值维持在合适的范围内。
6.为实现上述目的,本发明的实施例提供了壳聚糖/普鲁兰多糖微纤维载体,包括芯层以及形成与芯层外侧的壳层,芯层为壳聚糖与普鲁兰多糖的共混物,壳层为在芯层表面至少由壳聚糖与三聚磷酸钠(tpp)交联形成。
7.在本发明的一个或多个实施方式中,芯层的共混物中壳聚糖(cs)与普鲁兰多糖(pul)的质量比为(1-3):1。芯层的最佳的力学性能出现在cs/pul为2:1时。
8.在本发明的一个或多个实施方式中,壳聚糖与tpp的质量比为(3-9):(75-125)。
9.在本发明的一个或多个实施方式中,微纤维的直径为29μm-40μm。
10.在本发明的一个或多个实施方式中,壳聚糖/普鲁兰多糖微纤维载体的制备方法,包括如下步骤:准备芯层纺丝液和壳层纺丝液,芯层纺丝液为壳聚糖和普鲁兰多糖的混合溶液,壳层纺丝液为三聚磷酸钠溶液;采用同轴注射器实施微流纺丝,同轴注射器具有内流道以及形成与内流道外侧的外流道;微流纺丝时,芯层纺丝液输送至内流道,壳层纺丝液输送至外流道。
11.在本发明的一个或多个实施方式中,壳聚糖和普鲁兰多糖的混合溶液为至少由2-3wt.%的壳聚糖溶液与3-15wt.%的普鲁兰多糖溶液混合得到。优选的壳聚糖和普鲁兰多糖的混合溶液为至少由2-3wt.%的壳聚糖乙酸溶液与3-15wt.%的普鲁兰多糖水溶液混合得到。
12.在本发明的一个或多个实施方式中,壳聚糖和普鲁兰多糖的混合溶液为至少由质量比(1-4):1的壳聚糖溶液和普鲁兰多糖溶液混合得到。
13.在本发明的一个或多个实施方式中,三聚磷酸钠溶液的浓度为2-10wt.%。
14.在本发明的一个或多个实施方式中,微流纺丝时,芯层纺丝液和壳层纺丝液的流速比为(8-10):(25-30)。
15.在本发明的一个或多个实施方式中,cs/pul质量比为1:1、tpp浓度为2wt%时,壳聚糖/普鲁兰多糖微纤维载体的溶胀性能最佳,溶胀率达到227%。
16.在本发明的一个或多个实施方式中,缓释药物,包括如前述的壳聚糖/普鲁兰多糖微纤维载体以及形成于载体上的药物组分,药物组分选自对乙酰胺基酚、布洛芬、茶多酚、双氯芬酸、阿霉素、硫酸小檗碱、四环素。优选的,药物组分负载于芯层。
17.在本发明的一个或多个实施方式中,缓释药物中,药物组分的含量为4-10wt%。
18.在本发明的一个或多个实施方式中,缓释药物的制备方法,包括如下步骤:准备芯层纺丝液和壳层纺丝液,芯层纺丝液为壳聚糖、普鲁兰多糖和药物组分的混合溶液,壳层纺丝液为三聚磷酸钠溶液;采用同轴注射器实施微流纺丝,同轴注射器具有内流道以及形成与内流道外侧的外流道;微流纺丝时,芯层纺丝液输送至内流道,壳层纺丝液输送至外流道。
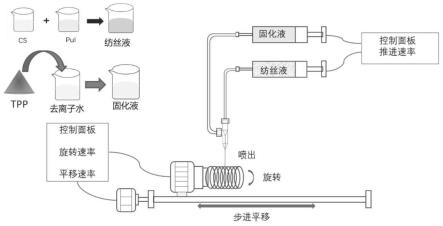
19.在本发明的一个或多个实施方式中,药物组合物,包括如前述的缓释药物。此时,药物组合物除了适量前述的缓释药物外,还可以视剂型选择性的添加糊精等片剂用辅料、生理盐水等液剂用辅料。
20.与现有技术相比,根据本发明实施方式的壳聚糖/普鲁兰多糖微纤维载体及其制备方法、缓释药物及制备方法、药物组合物,载体成型性好,生产加工性好,对药物负载能力强,并且具有良好的长效缓释性能。
附图说明
21.图1是根据本发明一实施方式的壳聚糖/普鲁兰多糖微纤维载体的制备流程示意图;
22.图2是根据本发明一实施方式的不同cs/pul质量比(tpp浓度为2wt%)的壳聚糖/普鲁兰多糖微纤维载体的sem图:(a)1:1;(b)2:1;(c)3:1;
23.图3是根据本发明一实施方式的不同cs/pul质量比(tpp浓度为2wt%)的壳聚糖/普鲁兰多糖微纤维载体的显微镜图:(a)1:1;(b)2:1;(c)3:1;
24.图4是根据本发明一实施方式的不同tpp浓度(cs/pul质量比为1:1)的壳聚糖/普鲁兰多糖微纤维载体的sem图:(a)2wt%(b)6wt%(c)10wt%;
25.图5是根据本发明一实施方式的不同tpp浓度(cs/pul质量比为1:1)的壳聚糖/普鲁兰多糖微纤维载体的显微镜图:(a)2wt%(b)6wt%(c)10wt%;
26.图6是根据本发明一实施方式的cs、pul、cp复合纤维的红外光谱图:(a)cs;(b)pul;(c)壳聚糖/普鲁兰多糖微纤维载体;
27.图7是根据本发明一实施方式的cs、pul的xrd图;
28.图8是根据本发明一实施方式的不同纺丝液组分的壳聚糖/普鲁兰多糖微纤维载体xrd图:(a-c)cs/pul为1:1时,tpp浓度分别为2wt%、6wt%、10wt%;(d-f)cs/pul为2:1时,tpp浓度分别为2wt%、6wt%、10wt%;(g-i)cs/pul为3:1时,tpp浓度为2wt%、6wt%、10wt%;
29.图9是根据本发明一实施方式的壳聚糖/普鲁兰多糖微纤维载体的力学分析图:(a)断裂强度;(b)断裂伸长率;
30.图10是根据本发明一实施方式的不同tpp浓度的壳聚糖/普鲁兰多糖微纤维载体在4h内的溶胀曲线:(a)2wt%;(b)6wt%;(c)10wt%;
31.图11是根据本发明一实施方式的不同cs/pul质量比的壳聚糖/普鲁兰多糖微纤维载体在4h内的溶胀曲线:(a)1:1;(b)2:1;(c)3:1;
32.图12是对乙酰氨基酚在磷酸盐缓冲溶液(ph=7.4)中的标准曲线;
33.图13是根据本发明一实施方式的不同tpp浓度的cpa(4wt%aap)复合纤维的sem图:(a)2wt%、(b)6wt%、(c)10wt%;
34.图14是根据本发明一实施方式的不同tpp浓度的cpa(4wt%aap)复合纤维的显微镜图:(a)2wt%、(b)6wt%、(c)10wt%;
35.图15是根据本发明一实施方式的不同aap含量的cpa(2wt%tpp)复合纤维的sem图:(a)4wt%、(b)7wt%、(c)10wt%;
36.图16是根据本发明一实施方式的不同aap含量的cpa(2wt%tpp)复合纤维的显微镜图:(a)4wt%、(b)7wt%、(c)10wt%;
37.图17是根据本发明一实施方式的cpa复合纤维的红外光谱图:(a)cs、(b)pul、(c)aap、(d)cpa复合纤维;
38.图18是根据本发明一实施方式的aap、cp复合纤维、cpa复合纤维的xrd图:(a)aap、(b)cp复合纤维、(c)cpa复合纤维;
39.图19是根据本发明一实施方式的cs,pul,cpa复合纤维的热重分析(tga)和微熵热重分析(dtg):(a)热重分析图(tga);(b)微熵热重分析图(dtg);
40.图20是根据本发明一实施方式的不同aap含量的cpa复合纤维的体外释药曲线:(a)4wt%、(b)7wt%、(c)10wt%;
41.图21是根据本发明一实施方式的不同tpp浓度制备的cpa复合纤维的体外释药曲线:(a)2wt%、(b)6wt%、(c)10wt%。
具体实施方式
42.下面结合附图,对本发明的具体实施方式进行详细描述,但应当理解本发明的保护范围并不受具体实施方式的限制。
43.除非另有其它明确表示,否则在整个说明书和权利要求书中,术语“包括”或其变换如“包含”或“包括有”等等将被理解为包括所陈述的元件或组成部分,而并未排除其它元件或其它组成部分。
44.本发明壳聚糖/普鲁兰多糖微纤维载体的制备主要包括,如图1所示:首先,称取一定质量的壳聚糖(cs)粉末溶于2wt%的乙酸溶液中,用磁力搅拌器搅拌至澄清溶液,静置、脱泡后得到适当浓度的cs溶液。其次,取一定质量的普鲁兰多糖(pul)粉末溶在去离子水中,搅拌至完全溶解,静置脱泡,得到适当浓度的pul溶液。然后将cs溶液和pul溶液按一定比例混合,常温下搅拌至溶液透明且无泡,作为芯层纺丝液。最后,称取一定质量的三聚磷酸钠(tpp)粉末溶解在去离子水中,得到一定浓度的tpp溶液。在常温常压下,用一支10ml注射器吸入一定量芯层纺丝液,再用一支20ml的注射器吸入一定量的壳层纺丝液tpp溶液,将两只注射器安装在注射泵上,在通过硅胶软管连接好注射器、同轴针头和接收装置。通过控制面板调节纺丝液和固化液的流速,设置滚筒接收器的转速,这样单根纤维就能在牵伸力的作用下缠绕到滚筒,然后设定循环周期数和平动速率,使得纤维在滚筒上有序地卷绕,得到排列整齐的cp复合纤维膜。
45.实施例1
46.本实施例的壳聚糖/普鲁兰多糖微纤维载体的制备为:3wt%的cs的乙酸溶液与15wt%的pul的去离子水溶液按照1:1的质量比进行共混得到芯层纺丝液,2wt%的tpp的去离子水溶液作为壳层纺丝液,设置微流体纺丝机参数为:滚筒接收装置的旋转速率为25r/min,平动速度1mm/min,芯层纺丝液流速8ml/h,壳层纺丝液流速25ml/h时,制备cp纤维,纤维成型良好,可形成均匀排列的纤维膜。
47.实施例2
48.本实施例的壳聚糖/普鲁兰多糖微纤维载体的制备为:3wt%的cs的乙酸溶液与15wt%的pul的去离子水溶液按照2:1的质量比进行共混得到芯层纺丝液,2wt%的tpp的去离子水溶液作为壳层纺丝液,设置微流体纺丝机参数为:滚筒接收装置的旋转速率为25r/min,平动速度1mm/min,芯层纺丝液流速8ml/h,壳层纺丝液流速25ml/h时,制备cp纤维,纤维成型良好,可形成均匀排列的纤维膜。
49.实施例3
50.本实施例的壳聚糖/普鲁兰多糖微纤维载体的制备为:3wt%的cs的乙酸溶液与15wt%的pul的去离子水溶液按照3:1的质量比进行共混得到芯层纺丝液,2wt%的tpp的去离子水溶液作为壳层纺丝液,设置微流体纺丝机参数为:滚筒接收装置的旋转速率为25r/min,平动速度1mm/min,芯层纺丝液流速8ml/h,壳层纺丝液流速25ml/h时,制备cp纤维,纤维成型良好,可形成均匀排列的纤维膜。
51.实施例4
52.本实施例的壳聚糖/普鲁兰多糖微纤维载体的制备为:2wt%的cs的乙酸溶液与3wt%的pul的去离子水溶液按照1:1的质量比进行共混得到芯层纺丝液,10wt%的tpp的去离子水溶液作为壳层纺丝液,设置微流体纺丝机参数为:滚筒接收装置的旋转速率为25r/
min,平动速度1mm/min,芯层纺丝液流速10ml/h,壳层纺丝液流速30ml/h时,制备cp纤维,纤维成型良好,可形成均匀排列的纤维膜。
53.实施例5
54.本实施例的壳聚糖/普鲁兰多糖微纤维载体的制备为:2wt%的cs的乙酸溶液与10wt%的pul的去离子水溶液按照1:1的质量比进行共混得到芯层纺丝液,10wt%的tpp的去离子水溶液作为壳层纺丝液,设置微流体纺丝机参数为:滚筒接收装置的旋转速率为25r/min,平动速度1mm/min,芯层纺丝液流速10ml/h,壳层纺丝液流速30ml/h时,制备cp纤维,纤维成型良好,可形成均匀排列的纤维膜。
55.实施例6
56.本实施例的壳聚糖/普鲁兰多糖微纤维载体的制备为:2.5wt%的cs的乙酸溶液与8wt%的pul的去离子水溶液按照1:1的质量比进行共混得到芯层纺丝液,10wt%的tpp的去离子水溶液作为壳层纺丝液,设置微流体纺丝机参数为:滚筒接收装置的旋转速率为25r/min,平动速度1mm/min,芯层纺丝液流速10ml/h,壳层纺丝液流速30ml/h时,制备cp纤维,纤维成型良好,可形成均匀排列的纤维膜。
57.实施例7
58.本实施例的壳聚糖/普鲁兰多糖微纤维载体的制备为:2.5wt%的cs的乙酸溶液与12wt%的pul的去离子水溶液按照1.5:1的质量比进行共混得到芯层纺丝液,4wt%、的tpp的去离子水溶液作为壳层纺丝液,设置微流体纺丝机参数为:滚筒接收装置的旋转速率为40r/min,平动速度4mm/min,芯层纺丝液流速9ml/h,壳层纺丝液流速26ml/h时,制备cp纤维,纤维成型良好,可形成均匀排列的纤维膜。
59.实施例8
60.本实施例的壳聚糖/普鲁兰多糖微纤维载体的制备为:2.2wt%的cs的乙酸溶液与8wt%的pul的去离子水溶液按照2.5:1的质量比进行共混得到芯层纺丝液,8wt%的tpp的去离子水溶液作为壳层纺丝液,设置微流体纺丝机参数为:滚筒接收装置的旋转速率为30r/min,平动速度3mm/min,芯层纺丝液流速8.5ml/h,壳层纺丝液流速28ml/h时,制备cp纤维,纤维成型良好,可形成均匀排列的纤维膜。
61.实施例9
62.本实施例的壳聚糖/普鲁兰多糖微纤维载体的制备为:2.7wt%的cs的乙酸溶液与6wt%的pul的去离子水溶液按照3:1的质量比进行共混得到芯层纺丝液,7.5wt%的tpp的去离子水溶液作为壳层纺丝液,设置微流体纺丝机参数为:滚筒接收装置的旋转速率为45r/min,平动速度2mm/min,芯层纺丝液流速9.5ml/h,壳层纺丝液流速29ml/h时,制备cp纤维,纤维成型良好,可形成均匀排列的纤维膜。
63.实施例11-19对应地与实施例1-9的区别分别仅在于:芯层纺丝液还对应地添加有4wt%的药物组分(可以选择布洛芬)、4wt%的药物组分(可以选择布洛芬)、4wt%的药物组分(可以选择布洛芬)、40wt%的药物组分(可以选择茶多酚)、40wt%的药物组分(可以选择茶多酚)、40wt%的药物组分(可以选择茶多酚)、5wt%的药物组分(可以选择阿霉素)、3wt%的药物组分(可以选择双氯芬酸)、8wt%的药物组分(可以选择四环素)。包括而不限于上述有关spa的实施例中药物组分可以选自aap、布洛芬、茶多酚、双氯芬酸、阿霉素、硫酸小檗碱、四环素等,这里对其组合形式就不一一举例。
64.以下对本发明方案的部分实施例样品性能进行测试比较,具体方案选自如下的组合所包含的不同情形:本实施例的壳聚糖/普鲁兰多糖微纤维载体的制备为:选自3wt%的cs的乙酸溶液与15wt%的pul的去离子水溶液按照1:1、2:1或3:1的质量比进行共混得到芯层纺丝液,选自浓度为2wt%、6wt%或10wt%的tpp的去离子水溶液作为壳层纺丝液,设置微流体纺丝机参数为:滚筒接收装置的旋转速率为25r/min,平动速度1mm/min,芯层纺丝液流速8ml/h,壳层纺丝液流速25ml/h时,制备cp纤维,纤维成型良好,可形成均匀排列的纤维膜。
65.纺丝液的组分对纤维成型的影响
66.在微流体纺丝过程中,纺丝液不仅受到微流泵的推动力,其自身的流体力学性能也至关重要,主要是由于微流体纺丝过程中纺丝通道会缩小到微米级别,在微观环境下其流体力学性能不可忽视。因此,纺丝液的浓度以及纺丝液的组分对纤维成型影响很大。同时,利用离子交联的原理对纤维固化,交联剂浓度对纤维成型也很重要。
67.由于cs溶液本身就有一定的黏度,并且随着溶液浓度的增加其流动性不断下降,因此,壳聚糖试纺浓度选取流动性相对较好的2wt%和3wt%。由于普鲁兰多糖易溶于水,流动性很好,考虑到纤维的主要组分对纤维成型的影响,配置了3wt%,10wt%,12wt%,15wt%四种不同浓度的pul溶液。cs溶液和pul溶液按照质量比1:1,2:1,3:1,4:1分别进行共混得到纺丝液作为同轴纺丝的芯层纺丝液,同时选用2wt%和10wt%的tpp溶液用作壳层纺丝液,进行纺丝。设计如表1所示方案为例进行对照:
68.表1微流体纺丝液组分的实验方案及纺丝结果
69.[0070][0071]
通过对照发现3wt%的cs溶液与15wt%的pul溶液按照质量比1:1、2:1、3:1、4:1共混作为纺丝液时,可与10wt%的tpp溶液进行纺丝,且纤维基本不会发生断头。但降低tpp溶液浓度(2wt%tpp溶液)后,3wt%的cs溶液与15wt%的pul溶液按照质量比4:1共混作为纺丝液时,纤维成型较差,因此选取cs/pul质量比为1:1、2:1、3:1,tpp浓度为2wt%、6wt%、10wt%,进行后续对照验证。
[0072]
纺丝工艺参数对纤维成型的影响
[0073]
微流体纺丝的纤维接收装置主要依靠滚筒接收和步进平移。利用同轴针头,在针尖处外相溶液包覆内向溶液,离子交联反应快速发生纤维成型,然后通过旋转电机提供牵伸力,将纤维从针尖牵伸出来卷绕到滚筒上,步进电机不断平动,形成竖向排列均匀的纤维膜。旋转电机的旋转速率会影响纤维成型,当旋转速度过快,提供的牵伸力大,纤维易被拉断,造成断头多,不连续。当旋转速度低时,牵伸力小,纤维会出现不能及时被卷绕而出现串珠情况,成型差。步进电机配合旋转电机不断平移,有利于形成均匀排列的纤维膜,然而步进会对纤维产生一个与牵伸力垂直的力,当这个力过大时也会造成纤维易断头的情况出现。纺丝液的流速是通过微流泵设置,流速过慢,在针尖处由于快速交联会形成液滴状固体堵塞针头,固化液流速慢,纤维难以交联成型。所以,有必要找到一个合适的工艺参数,才能得到成型良好且排列均匀的纤维。综合评估后以如表2所示为例进行对照。
[0074]
表2微流体纺丝工艺的对照方案及纺丝成型结果
[0075]
[0076][0077]
通过对比发现,当滚筒接收装置的旋转速率为25r/min,平移速度1mm/min,芯层溶液流速8ml/h,壳层溶液流速25ml/h时,纤维成型良好,可形成均匀排列的纤维膜。对比后发现,以上参数均可用于1:1,2:1,3:1的质量比进行共混得到纺丝液和2wt%,6wt%,10wt%的壳层纺丝液。
[0078]
纺丝液组分对纤维形貌的影响
[0079]
通过对纺丝液组分的比较,确定将3wt%的cs溶液与15wt%的pul溶液按照1:1、2:1、3:1的质量比混合作为芯层纺丝液,设置壳层纺丝液的tpp浓度为2wt%、6wt%和10wt%,相同的微流体纺丝参数下(滚筒接收装置旋转速率为25r/min,平动速度1mm/min,芯层纺丝液流速8ml/h,壳层溶液流速25ml/h)制备了cp复合纤维,探究纺丝液组分对纤维形貌的影响。
[0080]
(1)cs/pul质量比对纤维形貌的影响
[0081]
图2是不同cs/pul质量比制备的cp纤维的表面形貌照片。从图中可以看出,制备的cp纤维均成型良好,且随着纺丝液中壳聚糖含量的增加,纤维表面逐渐变得光滑。壳聚糖含量低时,纤维表面有纵向条纹,这主要是由于壳聚糖含量低时,壳聚糖与三聚磷酸钠交联不够紧密,内部空隙量增加,在干燥过程中收缩,形成凹槽。
[0082]
图3为cs/pul质量比分别为1:1,2:1,3:1时的cp纤维的光学显微镜图。cs/pul质量比分别为1:1,2:1和3:1的cp纤维的直径分别为34.4
±
5.17μm、34.92
±
4.45μm和36.15
±
6.17μm。cp纤维的直径随着cs/pul比例的变化略有变化,但总体直径变化均在误差范围内,说明cs/pul比例的变化对纤维直径的影响并不大。
[0083]
(2)tpp浓度对纤维形貌的影响
[0084]
图4和图5分别为cs/pul质量比为1:1,tpp浓度分别为2wt%,6wt%,10wt%的cp纤维sem图和光学显微镜照片。从图4中可以看出,不同交联剂浓度的cp纤维均成型良好,且随
着tpp浓度的增加,纤维表面细小凹槽数量增加,可能是因为在壳聚糖含量相对低时,tpp浓度增加,使得交联反应加剧,纤维内部没有较大空隙,使得干燥过程中形成较多细小凹槽。
[0085]
图5显示tpp浓度分别为2wt%,6wt%,10wt%的cp纤维的直径分别为32.44
±
3.59μm、34.4
±
5.17μm和35.68
±
3.12μm。cp纤维的直径随着tpp浓度的增加直径呈现出略微增加,这可能是因为tpp浓度增加,壳聚糖与tpp反应更加充分,纤维内部结构改变,与水分子的作用力加强,水分子难以彻底挥发,使得纤维直径变粗。
[0086]
cp复合纤维的红外光谱分析
[0087]
为了进一步确定纤维内部的分子结构,运用傅里叶红外光谱仪对cp纤维进行了表征。这里选用的cp复合纤维的纺丝液组分为:cs/pul质量比1:1,tpp浓度2wt%。
[0088]
如图6所示分别为cs粉末,pul粉末,cp复合纤维的红外光谱图。图中曲线a为cs的红外光谱,在3319cm-1处的宽峰对应的壳聚糖o-h和n-h的伸缩振动,1650cm-1处的峰对应的壳聚糖c=o的伸缩振动,1072cm-1处的峰对应的c-o的伸缩振动。图中曲线b为普鲁兰多糖的红外光谱,在3626cm-1处的尖峰对应的o-h的伸缩振动,3290cm-1处的宽峰对应的o-h的伸缩振动,1650cm-1处对应的峰为-oco-基团,1208cm-1处为c-h的振动峰,1072cm-1处为c-h的伸缩振动,在959cm-1,872cm-1,703cm-1处的吸收峰分别为α-(1,6)糖苷键,α-吡喃葡萄糖环和α-(1,4)糖苷键。如图5中曲线c所示,在cp复合微纤维的红外图谱中,1123cm-1和872cm-1的吸收峰明显增强,主要是因为壳聚糖与tpp交联成功,引入了-p=o和-p-o-c。最终通过红外光谱测试说明壳聚糖与tpp交联成功,而与普鲁兰多糖仅为物理共混。
[0089]
cp复合纤维的x射线衍射分析
[0090]
x射线衍射分析主要是用来测定材料的内部结构,通过图谱中峰的宽窄高低来判断材料内部的晶粒尺寸大小,会对材料的溶液渗透性能做出一定判断。因为药物释放载体应具有一定的溶液渗透性,而运用x射线衍射对制备的cp复合纤维进行表征。
[0091]
图7为普鲁兰多糖,壳聚糖的xrd曲线。从图中可以出,普鲁兰多糖粉末在2θ为18
°
处有一个较宽的峰,这表明普鲁兰多糖具有典型的非晶态结构,属于无定型粉末,内部分子排列无序。而壳聚糖粉末在2θ为20
°
处有一个衍射峰,属于壳聚糖的特征峰。
[0092]
图8为不同纺丝液组分下cp复合纤维的xrd图,从图中a-c可以发现,在2wt%tpp浓度下,cs溶液与pul溶液的质量比为1:1时,复合纤维仅有一个宽峰,且峰值与壳聚糖和普鲁兰的峰值比较略有下降,说明壳聚糖与普鲁兰多糖内部存在相互作用。通过图8中d-f可以看出,当tpp浓度增加,尖峰数量增加,同时峰强也有一定增加,说明复合纤维内部较大结晶的数量增加,可能是壳聚糖与三聚磷酸钠的交联程度有关。通过图8中a、d、g看出,壳聚糖的相对含量增加,壳聚糖与交联剂之间的反应程度不一,形成不同强度的尖峰,说明纤维内部有不同结晶结构的交联壳聚糖。
[0093]
cp复合纤维的力学性能分析
[0094]
图9为cp复合纤维的断裂强度和断裂伸长率柱状图。从柱状图中可以清晰地发现随着tpp浓度的上升,cp复合纤维的断裂强度逐渐增加的同时断裂伸长率逐渐下降,这可能是因为tpp浓度提高,在纤维固化过程中的交联反应程度增加,纤维内部结晶结构变化,断裂强度增大。随着壳聚糖相对含量的增加,当交联剂浓度为2wt%时,断裂强度相差不大,当交联剂浓度为6wt%,10wt%时,断裂强度先上升后下降。随着壳聚糖含量的增加,断裂伸长率呈现出先增大后减小。这也许与纤维中的氨基数目有关,壳聚糖含量提高,氨基数量增
加,使得分子间的氢键得到强化,力学性能变好,最佳的力学性能出现在cs/pul为2:1时,说明在这个比例下,壳聚糖的氨基和普鲁兰多糖的羧基之间能够形成有利的分子间氢键。当壳聚糖含量进一步提高时,壳聚糖形成分子内的氢键代替了分子间的氢键,使得力学性能下降。
[0095]
cp复合纤维膜的溶胀性能
[0096]
溶胀性能考察的就是纤维膜的吸水性能。若纤维膜具有较好的吸水性能,一方面可应用在创面伤口的敷料,能够很好的吸收组织液,保持创面清洁,减少感染。另一方面是基于药物释放系统,由于药物的释放其中的一个机理就是溶胀机理,纤维膜具有较好的溶胀性能,有利于药物的释放。
[0097]
在室温下,不同tpp浓度下制备的cp复合纤维膜在去离子水中的溶胀率随时间的变化曲线如图10所示。由图可以看出,随着时间的增加,cp纤维膜的溶胀率先增加后趋于稳定,基本所有的cp纤维膜在两个小时时达到溶胀平衡。tpp浓度为2wt%,cs/pul质量比为1:1的组分下制备的cp复合纤维膜的溶胀性能最佳,其溶胀率达到227%;而tpp浓度为10wt%,cs/pul质量比为3:1的组分下制备的cp复合纤维膜溶胀率最低,仅为73%。
[0098]
图11为室温下,不同cs/pul质量比的cp复合纤维膜在去离子水中的溶胀率随时间的变化曲线。从图中发现,在cs/pul质量比一样时,tpp浓度对cp复合纤维膜的溶胀性能的影响较大。从中发现随着交联剂浓度和cs/pul质量比的依次增加,cp纤维膜的溶胀率依次下降。主要是由于随着壳聚糖含量和交联剂浓度的变化,cp纤维内部结晶区含量变化,水分子越发难以渗透使得溶胀率变化。
[0099]
通过如上实施例可以知道:
[0100]
(1)3wt%的cs溶液与15wt%的pul溶液按照1:1、2:1或3:1的质量比进行共混得到芯层纺丝液,2wt%、6wt%或10wt%的tpp溶液作为壳层纺丝液,设置微流体纺丝机参数为:滚筒接收装置的旋转速率为25r/min,平动速度1mm/min,芯层纺丝液流速8ml/h,壳层纺丝液流速25ml/h时,制备cp纤维,纤维成型良好,可形成均匀排列的纤维膜。
[0101]
(2)形貌结构观察显示cs/pul质量比对纤维直径影响不大,cs/pul质量比分别为1:1、2:1和3:1的cp纤维的直径分别为34.4
±
5.17μm、34.92
±
4.45μm和36.15
±
6.17μm。tpp浓度为2wt%、6wt%、10wt%的cp纤维的直径分别为32.44
±
3.59μm、34.4
±
5.17μm和35.68
±
3.12μm,纤维直径随着tpp浓度的增加略微增加。不同cs/pul质量比或不同tpp浓度条件下纺制的cp纤维均成型良好,没有串珠结构。
[0102]
(3)cp纤维的傅立叶红外光谱(fitr)表明壳聚糖与tpp交联成功,与普鲁兰多糖仅为物理共混。xrd结果显示壳聚糖与普鲁兰多糖内部存在相互作用,且tpp浓度增加,复合纤维内部较大结晶的数量增加,生成不同结晶结构的复合纤维。
[0103]
(4)力学性能结果说明cp纤维的断裂强力随着tpp浓度的增加而增加,断裂伸长率逐渐下降。随着壳聚糖含量的增加,当tpp浓度为2wt%时,断裂强力相差不大,当tpp浓度为6wt%、10wt%时,断裂强度先增后减,断裂伸长率也随着壳聚糖含量的增加呈现出先增后减的趋势。
[0104]
(5)溶胀性能结果表明cp纤维膜在两小时达到溶胀平衡,提高tpp浓度或cs/pul质量比,cp纤维膜的溶胀率均下降。cs/pul质量比为1:1,tpp浓度为2wt%的cp纤维膜的溶胀性能最佳,溶胀率达到227%。
[0105]
进一步地载药性能进行验证:
[0106]
本发明缓释药物即载药的壳聚糖/普鲁兰多糖微纤维载体的制备可以为取一定质量的壳聚糖(cs)粉末加入2wt%乙酸溶液中,用磁力搅拌器搅拌至无颗粒,放置至无气泡,便得到适当浓度的cs溶液。其次,取一定质量的普鲁兰多糖(pul)粉末加入去离子水中,搅拌至溶解完全后停止搅拌,放置至无气泡,然后制得适当浓度的pul溶液。最后,将cs溶液和pul溶液1:1混合,用磁力搅拌器搅拌至混合均匀,再放置至无气泡,再称取一定重量的对乙酰氨基酚(aap)加入混合溶液,在37℃水浴锅中搅拌至无颗粒,得到cs/pul/aap混合溶液作为芯层纺丝液,并控制芯层纺丝液中aap的添加量占总溶质的4wt%、7wt%、10wt%。最后,取一定质量的三聚磷酸钠(tpp)固体粉末溶于去离子水中,得到适当浓度的tpp溶液作为壳层纺丝液。在常温常压下,用一支10ml注射器吸入一定量芯层纺丝液,再用一支20ml的注射器吸入一定量的壳层纺丝液tpp溶液,将两只注射器安装在注射泵上,在通过硅胶软管连接好注射器、同轴针头和接收装置。通过控制面板调节纺丝液和固化液的流速,设置滚筒接收器的转速,这样单根纤维就能在牵伸力的作用下缠绕到滚筒,然后设定循环周期数和平动速率,使得纤维在滚筒上有序地卷绕,得到排列整齐的cpa复合纤维膜。
[0107]
cpa复合纤维释药性能
[0108]
(1)对乙酰氨基酚(aap)标准曲线的标定
[0109]
称取适量的对乙酰胺基酚溶解在磷酸盐缓冲液(ph=7.4)中,利用型号为uv-5800的紫外分光光度计扫描配置好的对乙酰氨基酚溶液,扫描的波长范围为200-600nm,发现对乙酰胺基酚在磷酸盐缓冲液中的最大吸收波长为243nm,所以选择在243nm处测定对乙酰氨基酚溶液的吸光度来绘制标准曲线。
[0110]
称取5mg对乙酰氨基酚溶解在10ml的磷酸盐缓冲液(ph=7.4)中,将其移入50ml的容量瓶中进行定容,得到储备液。再用移液枪吸取0.1ml、0.2ml、1ml、2ml、4ml的储备液在10ml容量瓶中定容,利用型号为uv-5800的紫外分光光度计对已知浓度的标准溶液进行测定,得到药物在不同浓度时的吸光度大小,绘制对乙酰胺基酚标准曲线,计算线性相关系数。
[0111]
图12是对乙酰氨基酚在磷酸盐缓冲溶液(ph=7.4)中的标准曲线。通过线性拟合得到线性回归方程为y=0.06875x+0.03859(r2=0.99799)。因此,在1μg/ml-40μg/ml的浓度范围内,aap的浓度与其在243nm处的紫外吸光度具有很好的线性关系。
[0112]
(2)cpa复合纤维体外释放aap测定
[0113]
利用恒温振荡水浴锅模拟人体体液温度,不同的震荡速率模拟人体体液的波动,ph=7.4的磷酸盐缓冲液模拟人体体液,将载药纤维放置在此环境下,进行体外药物释放的模拟,具体操作如下:
[0114]
称取20mg的cpa复合纤维,装入透析袋内,然后放置到含有30ml磷酸盐缓冲液的离心管中,密封后放入恒温振荡水浴锅中振荡,设定温度为37℃,振荡速率30r/min。
[0115]
然后,分别在10min、20min、30min、40min、50min、60min、70min、80min、90min、105min、120min和150min处取3ml药物释放溶液,测定其在243nm处的吸光度,然后向离心管加入3ml空白的磷酸盐缓冲液,保持溶液量恒定,每种复合纤维的体外药物释放实验重复三次。最后根据aap的标准曲线方程计算出aap的浓度,再计算出aap的累计释放百分率,绘制出相应的药物释放曲线。
[0116]
药物释放百分率
[0117]qn
为从第1次到第n次取样时的药物释放百分率
[0118]
a为纤维中的药物总量
[0119]cn
为第n次取样时释放介质中的药物浓度
[0120]ci
为第i次取样时释放介质中的药物浓度
[0121]
载药cpa复合纤维的可纺性能
[0122]
微流纺丝条件为:滚筒接收装置的旋转速率为25r/min,平移速度1mm/min,芯层溶液流速8ml/h,鞘流层层溶液流速25ml/h成功制备了cpa复合纤维,同时药物的加入对纤维的成型并没有影响,在不同交联剂浓度下,纤维依旧可以很好成型。因此,通过sem与光学显微镜对制备的cpa复合纤维进一步表征,以确认tpp浓度与aap含量对纤维形貌的影响。
[0123]
(1)tpp浓度对cpa复合纤维形貌的影响
[0124]
图13是tpp浓度分别为2wt%、6wt%、10wt%的cpa纤维sem图。从图中可以看出,不同tpp浓度的cpa纤维均成型良好,且随着tpp浓度的增加,纤维表面逐渐光滑圆润。
[0125]
图14为aap含量为4wt%,tpp浓度分别为2wt%、6wt%,10wt%的cpa纤维的光学显微镜图,tpp浓度分别为2wt%、6wt%、10wt%的cpa纤维的直径分别为32.63
±
2.53μm,31.39
±
3.08μm和37.8
±
5.39μm。当tpp浓度为10wt%时,纤维直径变化较大,纤维变粗,这可能是因为交联剂浓度高时,壳聚糖与tpp反应更加充分,纤维内部结构改变,与水分子的作用力加强,水分子难以彻底挥发,使得纤维直径变粗。
[0126]
(2)aap含量对cpa纤维形貌的影响
[0127]
图15为tpp浓度为2wt%,aap含量分别为4wt%、7wt%、10wt%的cpa纤维sem图。从图中可以看出,不同aap含量的cpa纤维均成型良好,表面光滑,没有明显的结块或者晶体析出,说明对乙酰氨基酚被很好地负载到纤维中,且随着aap含量的增加,纤维逐渐圆润表面凹槽减少。
[0128]
图16为tpp浓度为2wt%,aap含量分别为4wt%、7wt%、10wt%的cpa纤维的光学显微镜图。aap含量分别为4wt%、7wt%、10wt%的cpa纤维的直径分别为33.04
±
3.98μm、32.7
±
4.11μm、32.63
±
2.53μm。cpa纤维的直径随着交联剂浓度的改变略有变化,但总体直径变化均在误差范围内,说明模型药物aap的含量的变化对纤维直径的影响并不大。
[0129]
cpa复合纤维的红外光谱分析
[0130]
图17是cs粉末、pul粉末、aap粉末、cpa复合纤维的红外光谱图。如图中a曲线所示,壳聚糖的红外光谱,在3304cm-1
处的宽峰对应壳聚糖o-h和n-h的伸缩振动,1421cm-1
处的特征峰属于ch2的弯曲振动,1021cm-1
处的峰为c-o的伸缩振动。曲线b对应普鲁兰多糖的红外光谱,在3356cm-1
处的宽峰对应的o-h的伸缩振动,在1549,1199cm-1
处为c-h的振动峰,1070cm-1
处为c-h的伸缩振动,在872cm-1
处的吸收峰是α-吡喃葡萄糖环的。曲线c为对乙酰氨基酚的红外光谱图,aap在3326cm-1
、1650cm-1
、1560cm-1
处的特征峰对应的是-oh,酰胺键上的-c=o和c=c骨架的伸缩振动,1432cm-1
处的特征峰对应的环呼吸振动峰。曲线d为cpa载药纤维的红外光谱,cpa载药纤维的红外光谱中发现了cs,pul和aap的特征吸收峰,并未发现新的吸收峰,说明aap与cs,pul之间是通过物理共混的形式聚集的,对药物的理化性质没有影响。
[0131]
cpa复合纤维的xrd分析
[0132]
在研究制备的载药微纤维的药物释放动力学时,药物在聚合物载体中的物理状态也是一个重要参数,药物的物理状态可能从分子分散(无定形)到明确的晶体结构不等。因此,对所制备的载药微纤维进行xrd分析,来表征药物在微纤维中的分散状态。
[0133]
图18为aap、cp复合纤维和cpa复合纤维的xrd曲线。图中曲线a为对乙酰氨基酚的xrd曲线,其在处12.11
°
、13.76
°
、15.57
°
、16.62
°
、18.2
°
、20.39
°
、23.47
°
、24.39
°
、26.39
°
、32.51
°
出现了典型的特征衍射峰。cp复合纤维仅有一个较大的宽峰,说明其内部属于无定型结构。与曲线a相比,cpa复合纤维的图谱近乎直线,在14.07
°
、18.21
°
、22.21
°
、25.86
°
处有很小的尖峰,说明cpa复合纤维中仍有aap结晶存在,但强度相对降低。cpa复合纤维的热重(tg)分析
[0134]
热重分析技术是研究聚合物高分子材料的热稳定性和热降解动力学,不同材料制备的药物载体的热稳定性也各尽不同。
[0135]
从图19中a中可以看出cs显示了两个阶段的重量损失,分别在30-100℃和200-350℃,第一阶段的重量损失主要壳聚糖样品中水分的蒸发,第二阶段主要还是cs自身的分解造成的,包括壳聚糖环的失水降解,分子链上部分基团的裂解。图19中a中pul的重量损失也分为两个阶段,分别在30-100℃和250-350℃,第一阶段是pul中残留的水分的蒸发,第二阶段是一个重量迅速下降的过程,这是普鲁兰多糖分子主链的降解导致的。图19中a中cpa的热重分析曲线表明cpa复合纤维也有两个失重区间,第一区间为30-250℃,主要是样品中残留水分的蒸发以及壳聚糖上氨基的裂解。第二区间为250-350℃,主要是壳聚糖主链大分子的降解以及普鲁兰多糖分子主链共价键的降解。从图19中b的dtg曲线中可以看出cpa纤维的热稳定性较cs和pul有所降低,主要还是因为在交联反应过程壳聚糖分子链段重排,其内部结构发生改变,导致热稳定性降低。
[0136]
cpa复合纤维膜中药物体外释放的研究
[0137]
图20为不同aap含量的cpa复合纤维中的体外释药曲线图,从图中可以看出交联剂tpp浓度为10wt%,aap含量为10wt%的纺丝组分下制备的cpa复合纤维在150min内药物的累计释放量最高为59.16%。从图20中a-c:可以看出,随着tpp浓度的增加,aap从cpa纤维中的药物释放百分比在逐渐减少。这可能是因为cpa复合纤维中壳聚糖相对含量一样时,tpp浓度增加,交联程度增加,纤维内部结构紧密,使得药物释放难度加大,药物释放百分率降低。随着cpa纤维中aap含量的增加,药物释放百分率也在逐渐增加。
[0138]
图21不同tpp浓度下制备的cpa复合纤维的体外释药曲线。从图21中可以看出,tpp低浓度时,aap含量为10wt%,7wt%时的药物释放百分率明显高于4wt%。这可能是因为tpp浓度低,药物含量多,交联不紧密,水分子易于渗透,药物释放易于释放,使得药物释放百分率相对较高。随着交联剂浓度的增加,药物含量高时依然具有较高的药物释放百分率。同时,可以看出药物在前30min释放过程接近直线,后续释放过程越发缓慢,主要是因为前期的释放主要是纤维表层的突释,后期是纤维内部的药物由内向外的扩散释放,释放速率减慢。
[0139]
cpa复合纤维膜中药物释放机理的影响
[0140]
影响药物释放的主要因素是载体的结构,因此对不同tpp浓度下制备的cpa复合纤维进行药物释放动力学模型拟合。然而药物缓释过程复杂,一种机理模型通常难以解释,本
研究选取了理想的零级释放模型、一级释放模型、weibull,higuchi、ritger-peppas、hixson-crowell模型来拟合cpa纤维的释药机理,发现cpa纤维的药物释放过程,结果见表3~表5。
[0141]
表3 2wt%tpp的cpa复合纤维释药曲线的药物释放动力学模型拟合
[0142][0143]
表4 6wt%tpp的cpa复合纤维释药曲线的药物释放动力学模型拟合
[0144][0145]
表5 10wt%tpp的cpa复合纤维释药曲线的药物释放动力学模型拟合
[0146][0147]
表3-5分别是tpp浓度为2wt%、6wt%、10wt%的cpa复合纤维药物释放曲线的药物释放动力学模型拟合。基于相关系数r2来判断吻合程度,tpp浓度为2wt%、6wt%、10wt%的cpa复合纤维的药物释放曲线与药物释放动力学模型与korsemeyer-peppas模型和higuchi模型较吻合。进一步比较发现,tpp浓度为2wt%、6wt%、10wt%的cpa复合纤维的药物释放
曲线与药物释放动力学模型korsemeyer-peppas模型更吻合,其拟合方程的斜率分别为0.40595、0.44931、0.45204。tpp浓度为2wt%与6wt%的拟合方程斜率均小于0.45,说明其释药机理更接近菲克扩散。tpp为10wt%的拟合方程斜率略大于0.45,说明药物释放的机理为非菲克扩散。交联剂浓度为2wt%、6wt%、10wt%的cpa复合纤维药物释放曲线的药物释放动力学模型也与higuchi模型较吻合,说明部分药物从纤维中疏水部分释放出来。综合所述,cpa复合纤维的药物释放主要有两种方式,第一种是纤维骨架溶胀,纤维中亲水部分快速溶解,药物通过菲克机制扩散出来,第二种是纤维骨架降解,药物从纤维内部疏水部分扩散出来,药物释放逐渐变慢。
[0148]
通过如上对cpa复合纤维的形貌和结构进行了表征,药物在ph值=7.4的pbs缓冲液中的释放行为及释药机理,得出以下结论:
[0149]
(1)3wt%的cs溶液与15wt%的pul溶液按照1:1的质量比混合,然后在混合溶液中加入4wt%、7wt%、10wt%的模型药物aap得到芯层纺丝液和2wt%、6wt%、10wt%的tpp溶液作为壳层纺丝液,设置滚筒接收装置的旋转速率为25r/min,平动速度1mm/min,芯层纺丝液流速8ml/h,壳层纺丝液流速25ml/h时,成功制备了cpa复合纤维,纤维成型良好,可形成均匀排列的纤维膜。
[0150]
(2)显微镜观察表明,模型药物aap的含量的改变对纤维直径没有明显的影响,aap含量分别为4wt%、7wt%、10wt%的cpa复合纤维的直径分别为33.04
±
3.98μm、32.7
±
4.11μm、32.63
±
2.53μm。tpp浓度分别为2wt%、6wt%、10wt%的cpa复合纤维的直径分别为32.63
±
2.53μm、31.39
±
3.08μm和37.8
±
5.39μm,tpp浓度为10wt%时,纤维直径变化较大,纤维变粗。sem图表明,不同aap含量的cpa复合纤维均成型良好,没有块状或者晶体附着在纤维表面,说明aap被很好地负载到纤维中,且随着aap含量的增加,纤维逐渐圆润表面凹槽减少。
[0151]
(3)通过对cpa复合纤维的内部结构分析,发现模型药物aap与cs,pul之间是以物理共混的方式结合,部分药物以结晶形式存在于纤维中。cpa复合纤维的热稳定性较cs和pul有所降低,主要还是因为在交联反应过程中,壳聚糖分子链段发生重排,其内部结构发生改变,导致热稳定性降低。
[0152]
(4)cpa复合纤维的药物释放百分率随着交联剂浓度的增加而降低,随着模型药物aap含量的增加而增加。cpa复合纤维在150min内的药物累计释放率可达到59.16%。
[0153]
(5)cpa复合纤维药物释放曲线的药物释放动力学模型与korsemeyer-peppas模型和higuchi模型较吻合,说明其药物释放主要有两种机制,第一种是溶胀机制,第二种是降解机制,药物从纤维内部疏水部分释放出来。
[0154]
前述对本发明的具体示例性实施方案的描述是为了说明和例证的目的。这些描述并非想将本发明限定为所公开的精确形式,并且很显然,根据上述教导,可以进行很多改变和变化。对示例性实施例进行选择和描述的目的在于解释本发明的特定原理及其实际应用,从而使得本领域的技术人员能够实现并利用本发明的各种不同的示例性实施方案以及各种不同的选择和改变。本发明的范围意在由权利要求书及其等同形式所限定。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1