靶向CD22的纳米制剂及其制备方法和应用
靶向cd22的纳米制剂及其制备方法和应用
技术领域
1.本技术属于纳米药物技术领域,尤其涉及一种靶向cd22的纳米制剂及其制备方法和应用。
背景技术:
2.目前,b细胞恶性肿瘤(如霍奇金淋巴瘤、非霍奇金淋巴瘤和白血病)患者的治疗主要依靠化疗和单克隆抗体,如抗cd20或cd19。然而,由于单克隆抗体治疗费用高昂且患者敏感性不同,化疗仍是一线治疗选择。虽然化疗药物的非特异性分布和全身毒性通常限制了它们的应用,但药物输送系统,尤其是靶向药物输送系统(tdds)提供了一个有利的平台,可以避免这些缺点。
3.b细胞淋巴瘤类型多样(包括2种霍奇金淋巴瘤和5种非霍奇金淋巴瘤),使得b细胞淋巴瘤的化疗方案复杂。此外,b细胞淋巴瘤类型可以相互转化,导致细胞表型多变,从而导致缺乏准确和通用的药物递送系统。因此,本领域技术人员亟待寻找一种能够准确靶向b细胞淋巴瘤的方法。
技术实现要素:
4.本技术的目的在于提供一种靶向cd22的纳米制剂,以应用于制备b细胞淋巴瘤药物。
5.为了完成上述发明目的,本技术提供了以下技术方案:
6.一种靶向cd22的纳米制剂,所述纳米制剂主要由多聚物、阳离子表面活性剂和聚唾液酸组成,所述多聚物与所述阳离子表面活性剂自组装形成纳米微粒,所述纳米微粒的表面修饰有所述聚唾液酸。
7.本技术所提供的纳米制剂,主要由多聚物、阳离子表面活性剂和聚唾液酸组成,其中,多聚物与阳离子表面活性剂自组装形成纳米微粒,多聚物用于形成纳米微粒的基体,为纳米制剂提供一个疏水内核,阳离子表面活性剂促进多聚物形成纳米微粒并维持微粒构象,同时,阳离子表面活性剂还提供阳离子,使得纳米微粒表面带正电荷,聚唾液酸在水溶液中带负电荷,如此,使得聚唾液酸能够通过静电作用吸附于纳米微粒表面。
8.经实验发现,本技术实施例所提供的纳米制剂具有靶向cd22的作用,经聚唾液酸表面修饰形成的上述纳米制剂可显著提高cd22阳性细胞对纳米诱导剂的吸收,吸收过程依赖于cd22介导,且不会在cd22阴性细胞中过度积累,而且,还能进一步提高icd诱导剂的icd作用,增加细胞凋亡率。另外,本技术的纳米制剂还可减少网状内皮系统的过早清除,可在体内长期使用,有利于提高icd诱导剂的药效。此外,本技术所提供的纳米制剂为固体,呈规则球形,在多种温度和ph环境下性能稳定,同时,具有响应肿瘤酸性环境的药物释放特性。
9.相应地,本技术还提供了一种上述纳米制剂的制备方法,包括以下步骤:
10.提供分散有多聚物的有机相以及分散有阳离子表面活性剂的水相,将所述有机相与所述水相进行第一混合处理,制得纳米微粒;
11.将聚唾液酸与所述纳米微粒在水溶液中进行第二混合处理,制得纳米制剂。
12.本技术所提供的上述纳米制剂的制备方法,首先,将分散有多聚物的有机相与分散有阳离子表面活性剂的水相进行第一混合处理,使得多聚物与阳离子表面活性剂在互相混合的过程中自组装形成纳米微粒;然后,将聚唾液酸与纳米微粒在水溶液中进行第二混合处理,使得聚唾液酸在与纳米微粒混合的过程中通过静电作用吸附于纳米微粒的表面,从而制得本技术的上述纳米制剂。
13.相应地,本技术还提供了前述纳米制剂或由上述制备方法制得的纳米制剂在制备b细胞淋巴瘤药物中的应用。
14.cd22是一种在b细胞上表达的特异性标志物,已被证实在b细胞淋巴瘤中显着上调,成为b细胞淋巴瘤的一个治疗靶点。本技术提供的纳米制剂具有靶向cd22的作用,有望于应用在制备b细胞淋巴瘤药物中,增强药物作用,促进诱导b细胞淋巴瘤的icd,提高肿瘤细胞凋亡率,以及延长药物作用时间,降低药物的毒副作用,具有良好的应用前景。
附图说明
15.为了更清楚地说明本技术实施例中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本技术的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
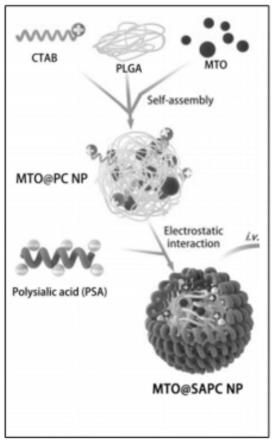
16.图1为实施例2的纳米药物的制备过程的简单示意图;
17.图2为sapc np、pc np、mto@sapc np和mto@pc np的tem图;
18.图3为sapc np、pc np、mto@sapc np和mto@pc np的化学表征结果,a为sapc np和pc np的平均流体动力学尺寸(average hydrodynamic size)检测结果,b为sapc np和pc np的流体动力学尺寸(hydrodynamic size)的分布情况,c为sapc np和pc np的ζ电位(ζ-potential)检测结果,d为mto@sapc np和mto@pc np的平均流体动力学尺寸(average hydrodynamic size)检测结果,e为mto@sapc np和mto@pc np的流体动力学尺寸(hydrodynamic size)的分布情况,f为mto@sapc np和mto@pc np的dlc(载药量)和ee(包封率)测试结果;
19.图4为mto@sapc np在不同环境下的稳定性测试结果,a、d的纵坐标为mto@sapc np的平均粒径,b、e的纵坐标为mto@sapc np的pdi,c的纵坐标为mto@sapc np的ζ电位,d、f的纵坐标为mto@sapc np的ζ电位;
20.图5为mto@sapc np在不同ph(5.5、6.4、7.4)的pbs中的药物释放曲线,其纵坐标为mto累积释放率;
21.图6为mto@pc np和mto@sapc np的细胞摄取行为的研究结果,c为各mto制剂与raji细胞孵育后的细胞clsm图像,f为各mto制剂与raji细胞孵育后细胞中的mto相对含量,其纵坐标为细胞内mto的相对含量;g为不同浓度的mto@sapc np与raji细胞孵育2小时后细胞中mto的荧光水平,其纵坐标为细胞内mto的相对荧光强度;
22.图7为不同mto制剂对淋巴瘤细胞活力的影响的检测结果,b为不同mto制剂对四种淋巴瘤细胞的半抑制浓度(ic50)检测结果,其纵坐标为mto制剂的ic50,横坐标中的每一细胞组从左到右分别对应的是mto、mto@pc np和mto@sapc np;d为不同mto制剂对raji的凋亡
率检测结果,纵坐标为raji细胞凋亡率;
23.图8为经不同mto制剂处理后的raji细胞的细胞线粒体膜电位(mmp)和ros水平的检测结果,b为细胞线粒体膜电位检测结果(mitochondrial membrane potential),d为ros是相对水平(relative level of ros);
24.图9为钙网蛋白(crt)在不同mto制剂处理后的raji细胞中的表达水平,e为经不同mto处理后的raji细胞进行染色后的共聚焦照片,f为raji细胞中crt表达水平的流式细胞图,g为raji细胞中crt表达水平的定量分析结果,其纵坐标为alexa488标记的crt单克隆抗体的荧光强度;
25.图10为经不同mto制剂处理后的raji细胞释放的hmgb1和atp水平的检测结果,h为raji细胞释放的hmgb1的相对含量的检测结果,纵坐标为细胞外hmgb1的相对含量;i为raji细胞的细胞外atp浓度的检测结果,纵坐标为atp浓度;
26.图11为使用流式细胞仪检测巨噬细胞(raw264.7细胞)用不同的mto制剂处理2小时后的mto荧光定量分析结果,纵坐标为细胞内mto的相对含量;
27.图12为注射不同mto制剂后的血药浓度分析结果,b为注射后72小时内的血浆中的mto浓度,c为与b对应的曲线下面积。
28.附图中,*表示p《0.05,**表示p《0.01,***表示p《0.001,ns表示差异无统计学意义。
具体实施方式
29.为了使本技术要解决的技术问题、技术方案及有益效果更加清楚明白,以下结合实施例,对本技术进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用于解释本技术,并不用于限定本技术。
30.一种靶向cd22的纳米制剂,所述纳米制剂主要由多聚物、阳离子表面活性剂和聚唾液酸组成,所述多聚物与所述阳离子表面活性剂自组装形成纳米微粒,所述纳米微粒的表面修饰有所述聚唾液酸。
31.多聚物主要为一类疏水的高分子聚合物,在本技术实施例中作为形成纳米微粒的基体,且为纳米制剂提供一个疏水内核。在一些实施例中,多聚物选自聚(乳酸-乙醇酸共聚物)、聚乳酸(pla)、聚己内酯(pcl)、聚氰基丙烯酸烷酯、聚酰胺-胺型树枝状高分子(pamam)中的至少一种。
32.阳离子表面活性剂为在水中能生成阳离子的表面活性剂,在使用传统的溶剂挥发方法制备纳米微粒的过程中,阳离子表面活性剂主要作为乳化剂来确保液滴的稳定性,直到液滴中的多聚物浓度足够高,从而形成纳米微粒并维持微粒构象。同时,阳离子表面活性剂还提供阳离子,使得纳米微粒表面带有正电荷,聚唾液酸在水溶液中带有负电荷,如此,使得聚唾液酸能够通过静电作用吸附于纳米微粒表面。在一些实施例中,阳离子表面活性剂选为十六烷基三甲基溴化铵。在具体实施例中,十六烷基三甲基溴化铵能够与聚(乳酸-乙醇酸共聚物)通过使用乳化-溶剂挥发法或透析法等方法自组装形成纳米微粒。
33.阳离子表面活性剂的用量影响着纳米微粒的形成及其微粒构象的稳定性。一些实施例中,纳米制剂中的阳离子表面活性剂的重量百分含量大于或等于1%。当控制纳米制剂中的阳离子表面活性剂的用量在该范围内,可促进形成呈规则球形、粒径均匀的纳米制剂。
34.聚唾液酸是唾液酸单体以α-2,8和/或α-2,9键连接的直链同聚物,其在水溶液中可形成阴离子,通过静电作用可吸附于所述纳米微粒的表面。一些实施例中,多聚物选为聚(乳酸-乙醇酸共聚物),阳离子表面活性剂选为十六烷基三甲基溴化铵,由此形成的纳米微粒通过与聚唾液酸相互作用,使得聚唾液酸均匀包覆在纳米微粒的表面,类似于以聚唾液酸为壳层、以纳米微粒为内核的核壳结构。
35.聚唾液酸的用量取决于纳米制剂中的阳离子表面活性剂的种类和用量,通过调节聚唾液酸相对于阳离子表面活性剂的用量,保证聚唾液酸能够均匀、稳定地吸附于纳米微粒的表面。一些实施例中,阳离子表面活性剂为十六烷基三甲基溴化铵,十六烷基三甲基溴化铵和聚唾液酸的重量比为1:(1-50)。
36.可以理解的是,本技术实施例所提供的纳米制剂实际上为具有纳米尺度的药物载体或药物制剂。当上述纳米制剂为具有纳米尺度的药物载体时,可简称为纳米载体。当上述纳米制剂为具有纳米尺度的药物制剂时,可简称为纳米制剂。
37.一些实施例中,纳米制剂还包括疏水性药物,该疏水性药物分散于纳米微粒中。进一步实施例中,该疏水性药物为能够引起肿瘤细胞免疫原性细胞死亡的化疗药物,化疗药物包括蒽环类化合物。在具体实施例中,化疗药物选为米托蒽醌。
38.在本技术实施例所提供的纳米制剂中,疏水性药物、多聚物、阳离子表面活性剂和聚唾液酸的相对用量应以能够形成纳米微粒、且聚唾液酸能修饰于纳米微粒的表面为前提来进行灵活调整。
39.一些实施例中,疏水性药物与多聚物的用量比为1:(1-100)。
40.在上述技术方案的基础上,本技术实施例以聚(乳酸-乙醇酸共聚物)、十六烷基三甲基溴化铵和聚唾液酸为原料,并调节各原料的具体用量,制得了一种纳米制剂,参考下文实施例1。对其进行化学表征,发现该纳米制剂为固体,呈规则球形,其粒径从100nm到700nm。
41.通过进一步对该纳米制剂进行性能研究,发现上述纳米制剂具有靶向cd22的作用,经聚唾液酸表面修饰形成的上述纳米制剂可显著提高cd22阳性细胞对纳米诱导剂的吸收,吸收过程依赖于cd22介导,且不会在cd22阴性细胞中过度积累。另外,当该纳米制剂负载了icd诱导剂(如米托蒽醌),其还进一步提高icd诱导剂的icd作用,增加细胞凋亡率。同时,本技术的纳米制剂还可减少网状内皮系统的过早清除,可在体内长期使用,有利于提高icd诱导剂的药效。此外,该纳米制剂在多种温度和ph环境下性能稳定,同时,具有响应肿瘤酸性环境的药物释放特性。
42.基于上述技术方案,本技术实施例还提供了上述纳米制剂的制备方法,其具体技术方案如下:
43.一种上述纳米制剂的制备方法,包括以下步骤:
44.s01、提供分散有多聚物的有机相以及分散有阳离子表面活性剂的水相,将所述有机相与所述水相进行第一混合处理,制得纳米微粒;
45.s02、将聚唾液酸与所述纳米微粒在水溶液中进行第二混合处理,制得纳米制剂。
46.具体地,在步骤s01中,分散有多聚物的有机相主要指的是将多聚物分散或溶解在有机溶剂中形成的多聚物溶液,分散有阳离子表面活性剂的水相主要指的是将阳离子表面活性剂分散或溶解在水中形成的阳离子表面活性剂水溶液。
47.通过将分散有多聚物的有机相与分散有阳离子表面活性剂的水相进行第一混合处理,从而使得多聚物与阳离子表面活性剂在互相混合的过程中自组装形成纳米微粒。将所述有机相与所述水相进行第一混合处理的步骤采用乳化-溶剂挥发法或透析法。
48.一些实施例中,将所述有机相与所述水相进行第一混合处理的步骤采用乳化-溶剂挥发法,一具体实施例为超声乳化-溶剂挥发法。该方法的具体操作过程可参考本领域的常规操作。可以理解的是,为促进多聚物与阳离子表面活性剂在混合体系中充分接触从而形成纳米微粒,有机相主要为中等极性或极性偏大、且易挥发的有机溶剂,包括但不限于甲醇、乙醇、乙腈等,使得在第一混合处理的过程中至少部分溶剂与水互溶。
49.一些实施例中,将所述有机相与所述水相进行第一混合处理的步骤采用透析法。可以理解的是,该方法中的有机相选为二甲基亚砜(dmso)。一具体实施例中,将分散有多聚物的有机相以及分散有阳离子表面活性剂的水相混合置于1000da的透析袋中,透析袋置于大量去离子水中,磁力搅拌48小时,每隔4小时更换去离子水,透析袋中的混悬液即为纳米微粒悬浮液。
50.当上述纳米制剂为具有纳米尺度的药物制剂时,所制备的纳米微粒中负载有疏水性药物。一些实施例中,有机相中还分散有疏水性药物,通过将疏水性药物和多聚物混合形成有机相,有利于促进疏水性药物均匀分散于纳米微粒中。
51.在步骤s02中,将聚唾液酸与纳米微粒在水溶液中进行第二混合处理,使得聚唾液酸在与纳米微粒混合的过程中通过静电作用吸附于纳米微粒的表面,从而制得本技术的上述纳米制剂。
52.可以理解的是,步骤s02中的水溶液指的是以水为溶剂的亲水体系,包括但不限于水、生理盐水、缓冲液等。
53.将聚唾液酸与所述纳米微粒在水溶液中进行第二混合处理的步骤采用机械搅拌的方法,使得两者能够在混合体系中充分接触即可。
54.另外,将聚唾液酸与纳米微粒在水溶液中进行第二混合处理的操作方法可参考本领域的常规操作,例如:将聚唾液酸与纳米微粒同时加入水中进行混合处理,或者,分别制得聚唾液酸水溶液和纳米微粒悬浮水溶液后进行混合处理,再或者,将聚唾液酸和纳米微粒之一制得水溶液后与另一混合处理。
55.cd22是一种在b细胞上表达的特异性标志物,已被证实在b细胞淋巴瘤中显着上调,成为b细胞淋巴瘤的一个治疗靶点,而前述纳米制剂或由上述制备方法制得的纳米制剂具有明显的靶向cd22的作用,由此,该纳米制剂有望于应用在制备b细胞淋巴瘤药物中。对该纳米制剂进行性能研究还发现,当该纳米制剂负载了icd诱导剂(如米托蒽醌),其还进一步提高icd诱导剂的icd作用,增加细胞凋亡率。同时,本技术的纳米制剂还可减少网状内皮系统的过早清除,可在体内长期使用,有利于提高icd诱导剂的药效。此外,该纳米制剂在多种温度和ph环境下性能稳定,同时,具有响应肿瘤酸性环境的药物释放特性。
56.综上,本技术实施例提供的纳米制剂具有靶向cd22的作用,将其用于制备b细胞淋巴瘤药物,有利于增强药物作用,促进诱导b细胞淋巴瘤的icd,提高肿瘤细胞凋亡率,以及延长药物作用时间,降低药物的毒副作用,具有良好的应用前景。
57.以下通过实施例对本发明的实施进行举例说明。
58.以下实施例中,聚(乳酸-乙醇酸共聚物)表示为plga(mw=7000da,乳酸与乙醇酸
的比例为75:25),十六烷基三甲基溴化铵表示为ctab,plga和ctab均购自sigma-aldrich(美国)。聚唾液酸表示为psa(colominic acid sodium salt,mw=30kda),购自carbosynth(china)。米托蒽醌表示为mto,购自med chem express(mce)。
59.实施例1
60.本实施例制备了一种纳米载体,多聚物选为plga,阳离子表面活性剂选为ctab,具体制备步骤如下:
61.s11、将30mg plga在超声波条件下溶解到乙腈中,作为有机相;称取10ml的1.5%ctab水溶液,作为水相;在10分钟内将有机相逐滴加到水相中,并在室温下继续超声搅拌,待有机溶剂蒸发后,通过0.45μm微孔膜过滤,收集滤液即为plga-ctab纳米微粒悬浮液;
62.s12、将plga-ctab纳米微粒悬浮液,在5分钟内滴加到1.5ml 1.5%psa水溶液中,连续搅拌6h后,用0.45μm微孔膜过滤得到聚唾液酸化纳米载体,标记为:sapc np。
63.对比例1
64.本对比例参照实施例1的步骤s11制得了plga-ctab纳米微粒,并以此作为对照用的纳米载体,标记为:pc np。
65.实施例2
66.参照图1,本实施例制备了一种纳米药物,多聚物选为plga,阳离子表面活性剂选为ctab,疏水性药物选为mto,具体制备步骤如下:
67.将30mg plga和3mg mto在超声波条件下溶解到乙腈中,作为有机相;称取10ml的1.5%ctab水溶液,作为水相;在10分钟内将有机相逐滴加到水相中,并在室温下继续超声搅拌,待有机溶剂蒸发后,通过0.45μm微孔膜过滤,收集滤液即为mto@plga-ctab纳米微粒悬浮液;
68.将mto@plga-ctab纳米微粒悬浮液,在5分钟内滴加到1.5ml 1.5%psa水溶液中,连续搅拌6h后,用0.45μm微孔膜过滤得到纳米药物,标记为:mto@sapc np。
69.对比例2
70.本对比例参照实施例2的步骤s21制得了mto@plga-ctab纳米微粒,并以此作为对照用的纳米药物,标记为:mto@pc np。
71.测试例
72.1、对sapc np、pc np、mto@sapc np和mto@pc np分别进行化学表征。
73.(1)将sapc np、pc np、mto@sapc np和mto@pc np以适当的浓度分散在水中,滴在铜网膜上。风干后,采用透射电子显微镜(tem,jem-1400显微镜,jeol,japan)进行形态观察并分析。
74.如图2所示,sapc np、pc np、mto@sapc np和mto@pc np为固体,呈规则球形,粒径基本相同,而且,sapc np、pc np、mto@sapc np和mto@pc np之间的形态没有显著差异。
75.(2)分别检测sapc np、pc np、mto@sapc np和mto@pc np的粒径、pdi、zeta电位、载药量和包封率,检测结果如图3所示。
76.sapc np和pc np的粒径从100nm到700nm,以250-300nm的粒径居多,如图3中的a所示,pc np的平均粒径为283
±
32nm,sapc np的平均粒径为292
±
41nm;如图3中的d所示,mto@pc np的平均粒径为293
±
22nm,mto@sapc np的平均粒径为300
±
31nm。
77.进一步分析各纳米制剂的流体动力学尺寸分布,结果如图3中的b和图3中的e所
示,进一步分析其pdi(多分散系数),pc np的pdi为0.11,sapc np的pdi为0.16,mto@pc np的pdi为0.16,mto@sapc np的pdi为0.17,均小于0.3,表明纳米载体具有均匀的粒径,综上数据,可以表明psa对pc np和mto@pc np的改性均不会影响材料的形貌、尺寸分布和平均尺寸。
78.图3中的c为sapc np和pc np的表面ζ电位检测结果,如图所示,pc np的ζ电位为7.46mv,由于带负电荷的psa对pc np的修饰,sapc np的表面ζ电位已从7.46mv反转至-14.45mv。
79.图3中的f为mto@sapc np和mto@pc np的dlc(载药量)和ee(包封率)测试结果,如图所示,尽管mto@sapc np的重量更大,但mto@sapc np的dlc和ee分别为8
±
1%和92
±
2%,略高于mto@pc np(dlc:7
±
1%,ee:84
±
1%)。究其原因,或由于psa的改性,使得纳米颗粒的结构更紧凑,这限制了药物的自由扩散。
80.(3)采用苯酚硫酸比色法检测mto@sapcnp中psa的含量,测得16.73%。采用酸碱滴定法检测pc np中ctab的含量,测得2.12%。
81.2、将mto@sapc np置于不同温度(4℃和37℃)和不同ph(5.5、6.4、8 7.4)环境中进行稳定性测试,如图4所示,mto@sapc np在不同温度和不同ph环境下的粒径、pdi和ζ电位都没有显着变化,表明本技术实施例提供的mto@sapc np具有良好的储存稳定性。
82.3、采用lc-ms分别测定了mto@sapc np在不同ph(5.5、6.4、7.4)的pbs中的药物释放情况,图5为测得的药物释放曲线,如图所示,mto@sapc np在ph5.5(模拟溶酶体环境)和ph6.4(模拟肿瘤组织微环境)下表现出更快的药物释放,这可能是由于增加的h
+
浓度削弱了psa和pc核心之间的相互作用导致的。因此,可以说明mto@sapc np具有响应肿瘤酸性环境的药物释放特性。
83.4、研究mto@pc np和mto@sapc np的细胞摄取行为。
84.(1)选择raji细胞作为实验细胞,将其分别与不同浓度的mto@pc np、mto@sapc np和αcd22+mto@sapc np(为αcd22+与mto@sapc np的混合物)进行孵育,各孵育2小时,然后采用共聚焦激光扫描显微镜观察细胞内mto荧光,以及对细胞内mto的荧光水平进行定量分析,结果如图6所示。
85.图6中的c为各mto制剂以640μg/ml的浓度与raji细胞孵育2小时后的细胞clsm图像,图中灰色部分代表mto,如图所示,mto@sapc np表现出比mto@pc np和αcd22+mto@sapc np更强的细胞摄取行为。其中,αcd22+mto@sapc np组内raji细胞上的cd22与αcd22+结合,使得raji细胞上的cd22活性受到抑制,在此基础上,αcd22+mto@sapc np仍表现出与mto@pc np相近似的细胞摄取行为,这表明细胞对mto@sapc np的摄取行为是由cd22介导的。
86.图6中的f为各mto制剂以640μg/ml的浓度与raji细胞孵育2小时后细胞中mto的荧光水平,以mto@pc np组的含量为1进行定量分析,与图6中的c所表现的结果一致,raji细胞明显吞噬更多的mto@sapc np(相当于mto@pc np的29.33倍)。值得注意的是,在与αcd22预孵育后,细胞内mto的荧光水平下降到与mto@pc np相似。
87.图6中的g为不同浓度的mto@sapc np与raji细胞孵育2小时后细胞中mto的荧光水平,结果显示,随着mto@sapc np浓度的增加,raji细胞中mto的荧光强度也成比例增加,这表明cd22介导的内吞作用呈剂量依赖性。但值得注意的是,当浓度超过640μg/ml时,raji细胞中mto的荧光强度不再增加,这也说明吞噬过程受cd22表达水平的限制。
88.综上,图6结果表明:psa修饰可以显着提高b细胞淋巴瘤细胞对纳米诱导剂的吸收,这个过程是cd22介导的;其次,mto@sapc np不会在cd22阴性细胞中过度积累,从而在一定程度上避免了潜在的脱靶效应。
89.5、选择两种b细胞淋巴瘤细胞(raji和ramos)和两种t细胞淋巴瘤细胞(hl-60和jurkat),采用mtt法分别测试不同mto制剂对淋巴瘤细胞活力的影响,检测结果如图7所示。
90.raji和ramos这两种b细胞淋巴瘤细胞上的cd22的表达水平显著高于hl-60和jurkat,故raji和ramos作为cd22阳性细胞(cd22+),hl-60和jurkat作为cd22阴性细胞(cd22-)。
91.图7中的b为不同mto制剂对四种淋巴瘤细胞的半抑制浓度(ic50)检测结果,在raji细胞中,mto和mto@pc np的ic50分别为0.52μm和0.43μm,mto@sapc np的ic50显着降低至0.15μm。在ramos细胞中,与其他两种mto制剂(1.76μm和1.63μm)相比,ramos细胞对mto@sapcnp(0.95μm)更敏感。对于jurkat细胞和hl-60细胞,虽然mto@sapc np的ic50均低于mto@pc np和游离mto,但没有显着差异。其中,相对于cd22阴性细胞,cd22阳性细胞对mto@sapc np更为敏感,尤其是raji细胞。这一结果清楚地显示了mto@sapcnp的细胞毒性为cd22依赖性细胞毒性。
92.为了进一步研究mto@sapc np对淋巴瘤细胞的毒性是cd22介导的,本实施例选择raji作为实验细胞,研究不同mto制剂对淋巴瘤细胞的凋亡作用。图7中的d为不同mto制剂对raji的凋亡率检测结果,如图所示,虽然mto或mto@pc np分别可以诱导36
±
2%或46
±
3%的raji细胞凋亡,但mto@sapc np更显着地增加了凋亡率,达到70
±
2%。
93.6、利用流式细胞仪研究mto@sapc np对诱导b细胞淋巴瘤细胞的免疫原性细胞死亡(icd)的促进作用
94.(1)采用tmre作为细胞线粒体膜电位(mmp)的探针,采用dcfh-da作为ros探针,检测分别经游离mto、mto@pc np、mto@sapc np处理后的raji细胞的mmp和ros水平,结果如图8所示。
95.如图8中的b所示,用游离mto和mto@pc np处理的raji细胞的mmp下降到空白对照组(control)的81
±
3%和71
±
3%,而mto@sapc np组显着降低至36
±
7%,这表明经mto@sapc np处理的raji细胞表现出更为明显的线粒体功能障碍。
96.如图8中的d所示,mto@sapc np处理的raji细胞的ros水平接近于空白对照组的6.47倍,分别是mto组的2.89倍和mto@pc np组的2.58倍,这表明经mto@sapc np处理的raji细胞表现出更为明显的细胞内氧化应激的爆发。
97.(2)研究钙网蛋白(crt)在游离mto、mto@pc np、mto@sapc np处理后的raji细胞中的表达水平,结果如图9所示。
98.图9中的e为经游离mto、mto@pc np、mto@sapc np处理后的raji细胞使用alexa488标记的crt单克隆抗体对细胞进行染色后的共聚焦照片,图中白色面积越大表示细胞表面暴露的crt越多。
99.图9中的f为raji细胞中crt表达水平的流式细胞图,g为raji细胞中crt表达水平的定量分析结果,如图所示,经mto@sapc np处理后,crt在raji细胞膜上的表达水平分别是游离mto和mto@pc np的2.87倍和2.21倍,这一结果表明mto@sapc np可明显促进raji细胞表达crt,crt作为“吃我”的信号,会发生膜易位,募集吞噬细胞和抗原呈递细胞消除它们并
呈递肿瘤抗原。
100.(3)使用hmgb1检测试剂盒和atp assay system bioluminescence detectionkit(promega)分别检测经不同mto制剂处理后的raji细胞释放的hmgb1和atp水平,检测结果如图10所示。
101.图10中的h为raji细胞释放的hmgb1的相对含量的检测结果,如图所示,以空白对照组的含量为1,mto@sapc np表现出比mto@pc np和游离mto更显著的hmgb1释放量。hmgb-1与树突状细胞(dc)膜上的tlr4结合,促进dc成熟,呈递抗原并激活细胞毒性t细胞,从而诱导机体产生抗肿瘤免疫反应。
102.图10中的i为raji细胞的细胞外atp浓度的检测结果,如图所示,经mto@sapc np处理的raji细胞的atp浓度明显高于mto@pc np和游离mto,atp会释放趋化因子以招募免疫细胞并激活nlrp3炎性体。
103.综上,经mto@sapc np处理的raji细胞表现出更为明显的细胞内氧化应激的爆发与线粒体功能障碍,同时,明显促进raji细胞表达crt以及显著增加hmgb1和atp的释放,虽然mto@pc np可以促进这几个指标的增加,但是,mto@pc np与游离mto之间并没有显著差异,由此,表明通过psa修饰,mto@sapc np可以诱导更高水平的icd。
104.7、图11为使用流式细胞仪检测巨噬细胞(raw264.7细胞)用不同的mto制剂处理2小时后的mto荧光定量分析结果,如图所示,raw264.7细胞吞噬的mto@sapc np仅为mto的45.4%和mto@pc np的36.9%。
105.8、构建基于raji细胞的b细胞淋巴瘤小鼠模型,在注射后72小时内,在预定时间点采集静脉血进行血药浓度分析,并计算其mto累积量,结果如图12所示。
106.图12中的b为不同mto制剂在淋巴瘤小鼠血浆中的药物浓度变化,如图所示,游离mto组和mto@pc np组血浆浓度随时间迅速下降,mto@sapc np组在0.5-4h内保持一定的稳定状态,然后逐渐下降。其中,2h时,游离mto、mto@pc np和mto@sapc np的药物浓度分别为10.2、33.1和56.6μg/ml。
107.图12中的c为不同mto制剂在淋巴瘤小鼠血浆中的mto累积量,如图所示,mto@sapc np分别是游离mto和mto@pc np的2.37倍和1.65倍。结合图11的结果,推测其原因或为通过psa表面修饰形成的mto@sapc np可以减少网状内皮系统的过早清除,表明该纳米药物可在体内长期使用。
108.以上所述仅为本技术的较佳实施例而已,并不用以限制本技术,凡在本技术的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本技术的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1