一种融合单细胞转录组的空间转录组生物组织亚结构解析方法
1.本发明涉及生物信息技术领域,更具体地,涉及一种融合单细胞转录组的空间转录组生物组织亚结构解析方法、系统及计算机可读存储介质。
背景技术:
2.随着生物信息学技术的快速发展,特别是转录组学和遗传学的研究改变了人们对癌症的认识。单细胞rna测序(scrna-seq)技术的进展可以让研究人员更清楚地了解肿瘤的细胞组成内部结构,通过scrna-seq技术研究分析肿瘤相关细胞,并根据细胞的分子谱将细胞的类型划分成更精细的细胞亚群。在scrna-seq技术中,聚类分析技术极为关键。现有的基于基因表达方法主要是用pearson相关系数、spearman相关系数等指标进行分析。细胞亚群形成了复杂的生态系统,它们之间的相互作用会影响肿瘤进展和治疗结果,但关于肿瘤相关细胞亚群相互作用的方式还没研究透彻。scrna-seq的缺陷在于对组织样本进行处理的时候失去了组织的空间背景(即细胞环境),而空间转录组测序可以同时获得细胞的空间位置信息和基因表达数据,更适合研究肿瘤基质中的细胞相互作用和空间基因表达。
3.目前空间转录组的技术主要有两种:基于ngs技术的方法和基于成像的方法(包括基于iss的和基于ish的)。
4.基于ngs技术的方法:2016年,空间转录组学(st)技术被提出,以获得空间分辨的全转录组信息。2018年底,st技术被进一步开发为10xvisium。10xvisium检测法在分辨率以及运行时间上都有改进。slide-seq利用放置在载玻片上的随机barcode(一种用于区分的编码)珠子来捕获mrna。在slide-seq方法发表后不久,出现了另一种使用更小的barcode珠子的技术——高分辨率空间转录组技术(hdst)。dbit-seq可在组织中使用确定性barcode进行空间组测序,该方法基于微流体的方法将barcode传递到组织玻片的表面,以实现10μm像素大小的分辨率。stereo-seq使用随机barcodedna纳米球沉积在阵列模式中,以实现纳米级分辨率。seq-scope已经实现了亚细胞分辨率的空间barcode,可以用来可视化核和细胞质转录。nanostringgeomxdsp技术是将数据的捕获放在了一个个圆形的感兴趣区域(roi)中,其将紫外线照射到roi上,释放可光裂解的基因标签以进行测序定量。在所有基于ngs的方法中,均为收集空间barcoderna并进行测序,其中测序数据的基本单元是reads(测序短片段)。每个测序短片段(reads)的barcode用于绘制空间位置,而测序reads的其余部分被映射到基因组,以识别转录源,共同生成一个基因表达矩阵。
5.基于ish(原位杂交)和基于iss(原位测序)的方法:
6.上述两类方法均是以图像处理生成基因表达矩阵。基于ish的方法是以ish技术为基础,通过互补荧光探针杂交检测目标序列。smfish利用多条短的寡核苷酸探针来靶向同一mrna转录本的不同区域。虽然smfish具有高灵敏度和亚细胞空间分辨率,但由于标准显微镜中光谱重叠的固有限制,它一次只能针对几个基因。seqfish是一种多路smfish方法,通过连续几轮杂交、成像和探针剥离,多次检测单个转录本,但既昂贵又耗时。为了弥补
seqfish的大量耗时,merfish技术于2015年被发布。这种技术可以鉴定单个细胞中数千种rna的拷贝数和空间定位。它利用组合标签、连续成像等技术来提高检测通量,并通过二进制barcode来抵消单分子标记和检测错误。
7.基于iss的方法是直接读出组织内转录本的序列。baristaseq是一种基于缺口填充挂锁的方法,其读取长度增加到15个碱基。starmap使用barcode挂锁探针,与靶标杂交,通过添加第二个引物,针对挂锁探针旁边的位点,避免了逆转录(rt)步骤。这种方法避免了cdna转换的效率障碍,并通过增加第二个杂交步骤来降低噪音。前面所提到的方法都是基于对靶标的先验知识,而fisseq是一种非靶标的方法,即捕获所有种类的rna,但非靶向扩增会导致光学拥挤和灵敏度降低。
8.为了提高空间数据的精度,在空间转录技术没有突破的情况下,整合多层面多维度的数据是一个可行的办法,两种或多种数据模态的计算集成可以更好地表征组织中的空间细胞类型组成和局部细胞状态,比如将scrna-seq数据与用空间转录组数据整合用于聚类分析,可以得到更精准的分类结果。
技术实现要素:
9.本发明提供了一种融合单细胞转录组的空间转录组生物组织亚结构解析方法、系统及计算机可读存储介质,提高了空间转录数据的聚类精度及单细胞数据分类效果。
10.本发明的首要目的是为解决上述技术问题,本发明的技术方案如下:
11.本发明第一方面提供了一种融合单细胞转录组的空间转录组生物组织亚结构解析方法,包括以下步骤:
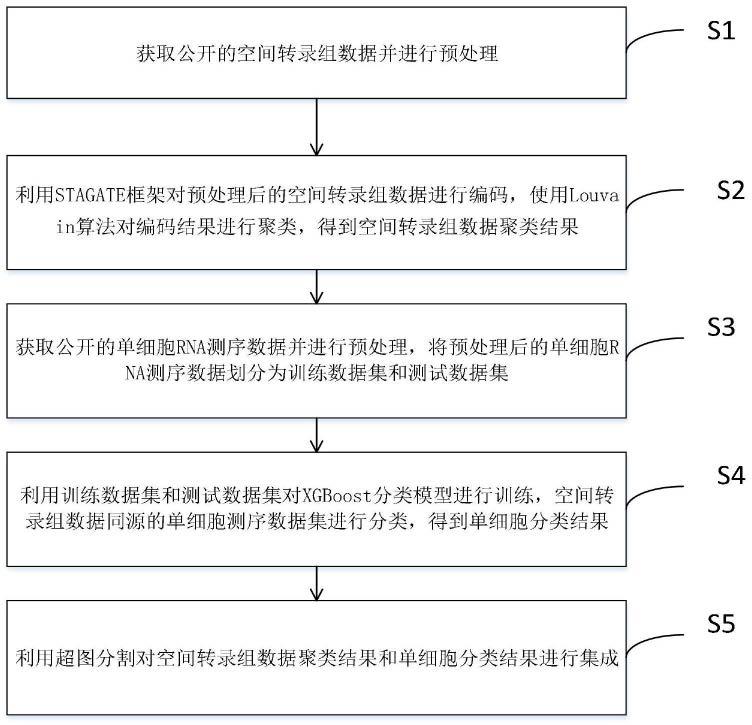
12.s1.获取公开的空间转录组数据并进行预处理;
13.s2.利用stagate框架对预处理后的空间转录组数据进行编码,使用louvain算法对编码结果进行聚类,得到空间转录组数据聚类结果;
14.s3.获取公开的单细胞rna测序数据并进行预处理,将预处理后的单细胞rna测序数据划分为训练数据集和测试数据集;
15.s4.利用训练数据集和测试数据集对xgboost分类模型进行训练,空间转录组数据同源的单细胞测序数据集进行分类,得到单细胞分类结果;
16.s5.利用超图分割对空间转录组数据聚类结果和单细胞分类结果进行集成。
17.进一步的,步骤s1中对公开的空间转录组数据并进行预处理包括:数据的归一化、数据格式调整。
18.进一步的,stagate框架包括:空间邻居网络snn和图注意力自动编码器,其中,空间邻居网络用于,所述图注意力编码器用于学习具有空间信息和基因表达的低维潜在向量embedding。
19.进一步的,空间邻居网络snn构建的具体过程为:
20.根据预定义的半径r将空间信息转换为无向邻居网络,定义a为snn的邻接矩阵,当且仅当节点i和节点j之间的欧几里德距离小于r时,a
ij
=1,a
ij
表示邻接矩阵a的第i行,第j列元素;对于其他不同技术的空间转录组数据,则根据数据的具体分辨率选择r,以每个节点为圆心,以r为半径,平均包含6-15个邻居节点;最后给每个节点添加自循环。
21.进一步的,图注意力自动编码器包括:编码器、解码器和图注意层,图注意力层嵌
在编码器与解码器中;
22.其中,编码器将节点的归一化的基因表达作为输入,并通过聚合该节点邻居的信息来生成节点向量spotembedding,编码器中的图注意力层共l-1层(k∈{1,2,...,l-1});
23.xi是节点i的归一化表达式,l是编码器的层数,为编码器第k层输出的节点向量spotembedding,si为节点s的邻居的集合,wk是可训练的权重矩阵;
24.将节点的表达谱作为初始节点向量spotembedding,则有:
[0025][0026]
其中是第k个图注意层输出中节点i和节点j之间的边权重;
[0027]
从节点i到其邻居节点j的边权重从节点i到其邻居节点j的边权重其中和是可训练的权重向量,sigmoid表示sigmoid激活函数;
[0028]
为了使空间相似性权重具有可比性,通过softmax函数对其进行归一化:即第k个图注意层输出中节点i和节点j之间的边权重;
[0029]
编码器第l层不采用注意力机制,输出为即最终输出的节点向量spotembedding;
[0030]
所述解码器在倒数第k层重构了节点i在第k-1层的embedding:1层的embedding:节点i在解码器最后一层的输出
[0031]
其中
[0032]
损失函数为
[0033]
进一步的,在对xgboost分类模型进行训练时,参数设置如下:设置预调整的参数学习率eta=0.7,迭代次数nround=20,节点分裂所需的最小损失函数下降值gamma=0.001,树的最大深度max_depth=5,最小样本权重的和min_child_weight=10。
[0034]
进一步的,利用超图分割对空间转录组数据聚类结果和单细胞分类结果进行集成具体步骤为:
[0035]
首先构成超图g,v代表所有基聚类的结果(簇)的集合,ci为其中的一个簇,e代表基于v构建的超边ei的集合,超边同时连接的点数大于或等于2个,每条超边中包含有多个节点,超边与超边之间包含的节点可以有重复,权重含的节点可以有重复,权重
[0036]
构成超图后,用mcla算法将图g分割成k个平衡的元簇类每个元簇类被一个表征示例和元簇类间的关联程度的m维指示向量元簇类被一个表征示例和元簇类间的关联程度的m维指示向量所表示,接下来将每个示例分配给与其最相关的元簇类,得到集成聚类簇λ,既优化后的最终聚类结果。
[0037]
本发明第二方面提供了一种融合单细胞转录组的空间转录组生物组织亚结构解析系统,该系统包括:存储器、处理器,所述存储器中包括一种融合单细胞转录组的空间转录组生物组织亚结构解析方法程序,所述一种融合单细胞转录组的空间转录组生物组织亚结构解析方法程序被所述处理器执行时实现如下步骤:
[0038]
s1.获取公开的空间转录组数据并进行预处理;
[0039]
s2.利用stagate框架对预处理后的空间转录组数据进行编码,使用louvain算法对编码结果进行聚类,得到空间转录组数据聚类结果;
[0040]
s3.获取公开的单细胞rna测序数据并进行预处理,将预处理后的单细胞rna测序数据划分为训练数据集和测试数据集;
[0041]
s4.利用训练数据集和测试数据集对xgboost分类模型进行训练,空间转录组数据同源的单细胞测序数据集进行分类,得到单细胞分类结果;
[0042]
s5.利用超图分割对空间转录组数据聚类结果和单细胞分类结果进行集成。
[0043]
本发明第三方面提供了一种计算机可读存储介质,所述计算机可读存储介质中包括融合单细胞转录组的空间转录组生物组织亚结构解析方法程序,所述融合单细胞转录组的空间转录组生物组织亚结构解析方法程序被处理器执行时,实现所述的一种融合单细胞转录组的空间转录组生物组织亚结构解析方法的步骤。
[0044]
与现有技术相比,本发明技术方案的有益效果是:
[0045]
本发明利用stagate转录组数据进行降维、分析与聚类,利用xgboost对单细胞转录数据聚,提高了空间转录数据的聚类精度及单细胞数据分类效果,同时利用,超图分割将两个聚类结果集成起来,获得精度更高的聚类结果。
附图说明
[0046]
图1为本发明一种融合单细胞转录组的空间转录组生物组织亚结构解析方法流程图。
[0047]
图2为本发明一种融合单细胞转录组的空间转录组生物组织亚结构解析系统框图。
具体实施方式
[0048]
为了能够更清楚地理解本发明的上述目的、特征和优点,下面结合附图和具体实施方式对本发明进行进一步的详细描述。需要说明的是,在不冲突的情况下,本技术的实施例及实施例中的特征可以相互组合。
[0049]
在下面的描述中阐述了很多具体细节以便于充分理解本发明,但是,本发明还可以采用其他不同于在此描述的其他方式来实施,因此,本发明的保护范围并不受下面公开的具体实施例的限制。
[0050]
实施例1
[0051]
如图1所示,本发明第一方面提供了一种融合单细胞转录组的空间转录组生物组织亚结构解析方法,包括以下步骤:
[0052]
s1.获取公开的空间转录组数据并进行预处理;
[0053]
需要说明的是,对转录组数据进行预处理包括:数据的归一化、数据格式调整。将
转录组数据进行归一化(筛选高差异基因等)、将数据格式转化为符合算法输入数据的格式。
[0054]
s2.利用stagate框架对预处理后的空间转录组数据进行编码,使用louvain算法对编码结果进行聚类,得到空间转录组数据聚类结果;
[0055]
需要说明的是,stagate框架包括:空间邻居网络snn和图注意力自动编码器,其中,空间邻居网络用于,所述图注意力编码器用于学习具有空间信息和基因表达的低维潜在embedding。stagate首先基于节点的相对空间位置构建空间邻居网络(snn),然后通过图注意力自动编码器学习具有空间信息和基因表达的低维潜在embedding(表示一个物体的一个抽象的向量),每个节点的归一化表达式先由编码器转换为d维潜在embedding,再通过解码器反转回重建的表达谱。其中,编码器和解码器的中间层采用了注意力机制,能自适应地学习snn的边缘权重(即相邻节点之间的相似性),并通过聚合某个节点的邻居的信息,用snn来更新该节点的表达。
[0056]
在一个具体的实施例中,空间邻居网络snn构建的具体过程为:
[0057]
根据预定义的半径r将空间信息转换为无向邻居网络,定义a为snn的邻接矩阵,当且仅当节点i和节点j之间的欧几里德距离小于r时,a
ij
=1,a
ij
表示邻接矩阵a的第i行,第j列元素。比如,对于10xvisium数据,我们将snn网络的半径r设置为能将每个节点的六个最近的节点包含进去的数值。对于其他不同技术的空间转录组数据,则根据数据的具体分辨率选择r,以每个节点为圆心,以r为半径,平均包含6-15个邻居节点;最后给每个节点添加自循环。
[0058]
图注意力自动编码器包括:编码器、解码器和图注意层,图注意力层嵌在编码器与解码器中;
[0059]
其中,编码器将节点的归一化的基因表达作为输入,并通过聚合该节点邻居的信息来生成节点向量spotembedding,编码器中的图注意力层共l-1层(k∈{1,2,...,l-1});
[0060]
xi是节点i的归一化表达式,l是编码器的层数,为编码器第k层输出的节点向量spotembedding,si为节点s的邻居的集合,wk是可训练的权重矩阵;
[0061]
将节点的表达谱作为初始节点向量spotembedding,则有:
[0062][0063]
其中是第k个图注意层输出中节点i和节点j之间的边权重;
[0064]
从节点i到其邻居节点j的边权重从节点i到其邻居节点j的边权重其中和是可训练的权重向量,sigmoid表示sigmoid激活函数;
[0065]
为了使空间相似性权重具有可比性,通过softmax函数对其进行归一化:即第k个图注意层输出中节点i和节点j之间的边权重;
[0066]
编码器第l层不采用注意力机制,输出为即最终输出的节点向量spotembedding;
[0067]
所述解码器在倒数第k层重构了节点i在第k-1层的embedding:1层的embedding:节点i在解码器最后一层的输出
[0068]
解码器的公式与编码器类似,其中
[0069]
损失函数为
[0070]
需要说明的是,本发明使用louvain算法对编码器输出对比结果(即节点向量spotembedding)进行聚类,得到空间转录组数据的聚类结果。其中louvain算法的分辨率是可以手动选择的,能够适应不同分辨率的空间转录组数据。
[0071]
s3.获取公开的单细胞rna测序数据并进行预处理,将预处理后的单细胞rna测序数据划分为训练数据集和测试数据集;
[0072]
需要说明的是,预处理包括数据进行归一化、调整数据格式等操作,将预处理后的单细胞rna测序数据划分为训练数据集和测试数据集。
[0073]
s4.利用训练数据集和测试数据集对xgboost分类模型进行训练,空间转录组数据同源的单细胞测序数据集进行分类,得到单细胞分类结果;
[0074]
需要说明的是,在对xgboost分类模型进行训练时,参数设置如下:设置预调整的参数学习率eta=0.7,迭代次数nround=20,节点分裂所需的最小损失函数下降值gamma=0.001,树的最大深度max_depth=5,最小样本权重的和min_child_weight=10。其中,如果训练后对分类精度不满意,可以在此基础上对参数进行相应的调整。最后使用训练后的模型对空间转录组数据同源(同一样本)的单细胞测序数据集进行分类,得到单细胞分类结果。
[0075]
s5.利用超图分割对空间转录组数据聚类结果和单细胞分类结果进行集成。
[0076]
需要说明的是,利用超图分割对空间转录组数据聚类结果和单细胞分类结果进行集成具体步骤为:
[0077]
首先构成超图g,v代表所有基聚类的结果(簇)的集合,ci为其中的一个簇,e代表基于v构建的超边ei的集合,超边同时连接的点数大于或等于2个,每条超边中包含有多个节点,超边与超边之间包含的节点可以有重复,权重节点,超边与超边之间包含的节点可以有重复,权重
[0078]
构成超图后,用mcla算法将图g分割成k个平衡的元簇类每个元簇类被一个表征示例和元簇类间的关联程度的m维指示向量元簇类被一个表征示例和元簇类间的关联程度的m维指示向量所表示,接下来将每个示例分配给与其最相关的元簇类,得到集成聚类簇λ,既优化后的最终聚类结果。
[0079]
如图2所示,本发明第二方面提供了一种融合单细胞转录组的空间转录组生物组织亚结构解析系统,该系统包括:存储器、处理器,所述存储器中包括一种融合单细胞转录组的空间转录组生物组织亚结构解析方法程序,所述一种融合单细胞转录组的空间转录组生物组织亚结构解析方法程序被所述处理器执行时实现如下步骤:
[0080]
s1.获取公开的空间转录组数据并进行预处理;
[0081]
s2.利用stagate框架对预处理后的空间转录组数据进行编码,使用louvain算法
对编码结果进行聚类,得到空间转录组数据聚类结果;
[0082]
s3.获取公开的单细胞rna测序数据并进行预处理,将预处理后的单细胞rna测序数据划分为训练数据集和测试数据集;
[0083]
s4.利用训练数据集和测试数据集对xgboost分类模型进行训练,空间转录组数据同源的单细胞测序数据集进行分类,得到单细胞分类结果;
[0084]
s5.利用超图分割对空间转录组数据聚类结果和单细胞分类结果进行集成。
[0085]
本发明第三方面提供了一种计算机可读存储介质,所述计算机可读存储介质中包括融合单细胞转录组的空间转录组生物组织亚结构解析方法程序,所述融合单细胞转录组的空间转录组生物组织亚结构解析方法程序被处理器执行时,实现所述的一种融合单细胞转录组的空间转录组生物组织亚结构解析方法的步骤。
[0086]
显然,本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1