一种全外显子组测序数据的处理方法、系统及一种检测短串联重复疾病相关异常扩增的系统与流程
1.本发明涉及医药技术领域,尤其涉及一种全外显子测序数据的处理方法、系统及一种检测短串联重复疾病相关异常扩增的系统。
背景技术:
2.基因组上外显子占总序列约1-2%,却包含高达85%疾病相关致病性变异。通过序列捕获或靶向技术将全基因组外显子区域dna富集后的高通量测序方法叫全外显子组测序(wes),因其全面性、有效性和极高性价比已经成为大多数临床异质性较高疾病的分子诊断首选方案,可以同时检测snp、indel、cnv,若增加线粒体环的捕获,还可以同时检测线粒体环基因变异。
3.短串联重复(str),通常指基因组中由1~6个碱基单元(motif)组成的一段dna重复序列。重复数在个体间高度变异且数量众多,具有丰富的遗传多态性,据估计在人类基因组中有超过一百万str位点,,占人类基因组的约3%。短串联重复扩增能够导致一系列疾病包括亨廷顿、各种共济失调、肌萎缩性侧索硬化症、额颞叶痴呆、脆x综合征和其他神经系统疾病。同时也有较多的研究表明串联重复多态性(trp)在多基因疾病的基因表达调控中发挥重要的作用。串联重复相关疾病(trds)在表型上并不是一个简单有和无的定义(患病人群和正常人群的比较),因其特殊性根据其串联重复次数的变化通常导致一种连续的量级的变化(比如发病的年龄、疾病严重程度等)。
4.目前,对于这类疾病的常规分子诊断是基于精确的pcr扩增或者southern印迹分析,这需要实验室能精准的扩增每种不同的重复序列,临床医生需要对患者进行准确的诊断,确定最可能与哪些疾病最相关,提交合适的检测。但是str相关疾病在临床症状、外显率的变异和发病时间上有一定的重叠,主要取决于等位基因的大小和修饰基因的作用。在多达50%的共济失调的患者中,可能是由其它突变比如snp、indel等导致。因此在对这些疾病进行分子诊断时,通常还需要对候选基因进行常规测序,比如panel、wes等。有些遗传病因临床表型异质性超高,可能因错误诊断而选择不恰当的检测方法而导致患者无法获得正确的分子诊断确诊。比如齿状核红核苍白球路易体萎缩症(dentatorubral-pallidoluysian atrophy,drpla))是一种进行性常染色体显性遗传疾病,其特征是肌阵挛性癫痫、共济失调、舞蹈手足徐动症/肌张力障碍、认知障碍、痴呆和精神障碍,由atn1基因cag三核苷酸串联重复导致,正常人重复次数为7-23次,受累者常为49-88次。drpla发病年龄从0岁到70岁,平均发病年龄为30岁。该病临床表现因发病年龄而异:儿童的主要特征是共济失调、智力障碍、行为改变、肌阵挛和癫痫;成人的主要特征是共济失调、舞蹈手足徐动症和痴呆。20岁之前发病的患者通常有进行性肌阵挛性癫痫((pme))表型,其特征为肌阵挛、癫痫发作、共济失调和进行性智力退化,还观察到各种形式的全面性癫痫发作((包括强直、失张力、阵挛或强直-阵挛发作))。对于早发型病人常常因被诊断为癫痫发作、智力障碍而推荐做常规wes检测。
5.虽然高通量测序技术(ngs)的发展为全基因组检测数以百万计的str提供了可能性,但是在生信分析中基因分型仍然具有挑战:高gc含量、无法覆盖完整重复的短读长序列、映射到与参考基因组存在差异的大的缺失/插入的str变异、重复序列本身重复特性无法映射或者映射差、pcr扩增导致的stutter产物(影子带或者dna聚合酶滑脱产物)噪音影响等。虽然illumina开发了一个免扩增的(pcr-)的文库制备方法,该方法去除了样本制备(pcr+)中pcr扩增过程中str stutter错误,可以提高str分型的准确性。但是,现阶段pcr+方法已经产生了大量的测序数据,pcr-的方法在成本和难度上还存在一定的限制。虽然pcr-的wgs测序有很多的优势,但是wes因其低成本高覆盖,在人类遗传病研究和诊断中发挥着重要的作用。在国内遗传诊断中,wes是临床异质性高的疾病首选的检测方式,因此,从pcr+的测序数据中精确的进行str分型至关重要。针对wes数据,现有技术开发了较多用于str分型的工具,但绝大多数限于检测读长范围内的str,而且因其算法和原理不同,在疾病相关str鉴定上都存在一定的局限性。比如exstra,该算法主要用于在测序的队列样本中检测用户指定的str序列,其为outlier检测手段,假设大多数(》85%)的个体在特定的str位点具有正常的等位基因。;又如expansionhunter,主要用于wgs数据的str分析,倾向于pcr-的文库制备,使用预定的阈值来确定个体是否存在str扩增。
6.目前ngs短读长测序数据的str分析的相关研究更多集中在分析算法上,但不同的算法、不同的测序平台、不同的探针、不同的比对软件对最终str分析结果都存在较大的影响,导致绝大多分子诊断送检样本均会存在分析软件提示的不同程度的异常值。
技术实现要素:
7.有鉴于此,本发明提供了一种全外显子测序数据的处理方法、系统及一种检测短串联重复疾病相关异常扩增的系统,本发明提供的全外显子测序数据的处理方法受不同的算法、不同的测序平台、不同的探针、不同的比对软件影响较小,得到的数据结果较为准确。
8.本发明提供了一种全外显子测序数据的处理方法,包括以下步骤:
9.步骤s1、获取第一参考数据,所述第一参考数据包括参考样本的str相关疾病基因靶标区域内碱基覆盖度数据和预定的碱基覆盖度下的样本占比数据;
10.获取第二参考数据,所述第二参考数据包括阴性参考样本扩增次数数据;
11.步骤s2、获取检测样本数据,所述检测样本数据包括检测样本的str相关疾病基因靶标区域内碱基覆盖度数据、预定的碱基覆盖度下的样本占比数据和检测样本扩增次数数据;
12.将所述检测样本的str相关疾病基因靶标区域内碱基覆盖度数据和预定的碱基覆盖度下的样本占比数据与所述参考样本的str相关疾病基因靶标区域内碱基覆盖度数据和预定的碱基覆盖度下的样本占比数据进行比对,获得第一比对结果数据;
13.若所述第一比对结果数据为不符合,则将所述检测样本扩增次数数据与所述阴性参考样本扩增次数数据进行比对,获得第二比对结果数据。
14.在一些实施例中,所述步骤s1具体包括:
15.获取str相关疾病基因数据,确定str相关疾病异常扩增的靶标区域数据;
16.获取参考样本的wes测序数据,对比所述参考样本的wes测序数据与所述str相关疾病异常扩增的靶标区域数据,获得参考样本的str相关疾病基因靶标区域内碱基覆盖度
数据和预定的碱基覆盖度下的样本占比数据;
17.采用expansionhunter软件对参考样本中阴性样本的wes测序数据进行分析,获得阴性参考样本扩增次数数据。
18.在一些实施例中,所述步骤s2具体为:
19.获取检测样本的wes测序数据,对比所述检测样本的wes测序数据与所述str相关疾病异常扩增的靶标区域数据,获得检测样本的str相关疾病基因靶标区域内碱基覆盖度数据和预定的碱基覆盖度下的样本占比数据;
20.采用expansionhunter软件对检测样本的wes测序数据进行分析,获得检测样本扩增次数数据。
21.在一些实施例中,所述步骤s1还包括:对所述阴性参考样本扩增次数数据进行修正,获得修正后的阴性参考样本扩增次数数据。
22.在一些实施例中,对所述阴性参考样本扩增次数数据进行修正具体为:
23.获取阳性样本的实际扩增次数数据和wes测序数据;
24.采用expansionhunter软件对阳性样本的wes测序数据进行分析,获得阳性样本的预测扩增次数数据;
25.根据所述阳性样本的实际扩增次数数据和预测扩充次数,对所述阴性参考样本扩增次数数据进行修正。
26.在一些实施例中,还包括:
27.采用exstra软件对所述参考样本的wes测序数据进行分析,获得所述参考样本中阳性样本的str计算得分;
28.采用exstra软件对所述检测样本的wes测序数据进行分析,获得所述检测样本的str计算得分。
29.本发明还提供了一种全外显子测序数据的处理系统,包括第一参考数据获取单元,所述第一参考数据单元用于获取第一参考数据,所述第一参考数据包括参考样本的str相关疾病基因靶标区域内碱基覆盖度数据和预定的碱基覆盖度下的样本占比数据;
30.第二参考数据获取单元,所以第二参考数据获取单元用于获取第二参考数据,所述第二参考数据包括阴性参考样本扩增次数数据;
31.检测样本数据获取单元,所述检测样本数据获取单元用于获取检测样本数据,所述检测样本数据包括检测样本的str相关疾病基因靶标区域内碱基覆盖度数据、预定的碱基覆盖度下的样本占比数据和检测样本扩增次数数据;
32.第一比对单元,所述第一比对单元用于将所述检测样本的str相关疾病基因靶标区域内碱基覆盖度数据和预定的碱基覆盖度下的样本占比数据与所述参考样本的str相关疾病基因靶标区域内碱基覆盖度数据和预定的碱基覆盖度下的样本占比数据进行比对,获得第一比对结果数据;
33.第二比对单元,所述第二比对单元用于比对所述检测样本扩增次数数据与所述阴性参考样本扩增次数数据,获得第二比对结果数据。
34.在一些实施例中,所述第一参考数据获取单元包括str相关疾病基因数据获取单元,所述str相关疾病基因数据获取单元用于获取str相关疾病基因数据,确定str相关疾病异常扩增的靶标区域数据;
35.参考样本wes测序数据获取单元,所述参考样本wes测序数据获取单元用于获取参考样本的wes测序数据;
36.第三比对单元,所述第三比对单元用于对比所述参考样本的wes测序数据与所述str相关疾病异常扩增的靶标区域数据,获得参考样本的str相关疾病基因靶标区域内碱基覆盖度数据和预定的碱基覆盖度下的样本占比数据。
37.在一些实施例中,所述检测样本数据获取单元包括检测样本wes测序数据获取单元,所述检测样本wes测序数据获取单元用于获取检测样本的wes测序数据;
38.第四比对单元,所述第四比对单元用于对比所述检测样本的wes测序数据与所述str相关疾病异常扩增的靶标区域数据,获得检测样本的str相关疾病基因靶标区域内碱基覆盖度数据和预定的碱基覆盖度下的样本占比数据;
39.检测样本扩增次数数据处理单元,用于采用expansionhunter软件对检测样本的wes测序数据进行分析,获得检测样本扩增次数数据。
40.本发明还提供了一种检测短串联重复疾病相关异常扩增的系统,包括第一参考数据获取单元,所述第一参考数据单元用于获取第一参考数据,所述第一参考数据包括参考样本的str相关疾病基因靶标区域内碱基覆盖度数据和预定的碱基覆盖度下的样本占比数据;
41.第二参考数据获取单元,所以第二参考数据获取单元用于获取第二参考数据,所述第二参考数据包括阴性参考样本扩增次数数据;
42.检测样本数据获取单元,所述检测样本数据获取单元用于获取检测样本数据,所述检测样本数据包括检测样本的str相关疾病基因靶标区域内碱基覆盖度数据、预定的碱基覆盖度下的样本占比数据和检测样本扩增次数数据;
43.第一比对单元,所述第一比对单元用于将所述检测样本的str相关疾病基因靶标区域内碱基覆盖度数据和预定的碱基覆盖度下的样本占比数据与所述参考样本的str相关疾病基因靶标区域内碱基覆盖度数据和预定的碱基覆盖度下的样本占比数据进行比对,获得第一比对结果数据;
44.第二比对单元,所述第二比对单元用于比对所述检测样本扩增次数数据与所述阴性参考样本扩增次数数据,获得第二比对结果数据;
45.预测系统,所述预测系统用于根据第一比对结果数据和第二比对结果数据,获得短串联重复疾病相关异常扩增的预测结果。
46.本发明通过wes测序数据中实际样本真实覆盖度情况定义样本可检测的str相关基因,比使用wes探针bed区域/bed+flanking区域的是否重叠来评估更准确。本发明提供的全外显子测序数据的处理方法受不同的算法、不同的测序平台、不同的探针、不同的比对软件影响较小,得到的数据结果较为准确。
附图说明
47.图1为本发明实施例提供的检测短串联重复疾病相关异常扩增的流程示意图;
48.图2为本发明实施例获取str相关疾病基因靶标区域内覆盖度情况的流程示意图;
49.图3为str疾病相关基因正常扩增范围内实际观测值和软件预测值;
50.图4为致病性str变异过滤指标和评估标准流程示意图。
具体实施方式
51.本发明提供了一种全外显子测序数据的处理方法、系统及一种检测短串联重复疾病相关异常扩增的系统。本领域技术人员可以借鉴本文内容,适当改进工艺参数实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明。本发明的方法及应用已经通过较佳实施例进行了描述,相关人员明显能在不脱离本发明内容、精神和范围内对本文的方法和应用进行改动或适当变更与组合,来实现和应用本发明技术。
52.参见图1,图1为本发明实施例提供的检测短串联重复疾病相关异常扩增的流程示意图,具体而言,本发明实施例提供的检测短串联重读疾病相关异常扩增方法包括以下步骤:
53.1)str相关疾病的列表、wes产品的靶标区域内覆盖度情况获取:
54.首先进行str相关罕见遗传病基因变异信息收集和实际生产数据样本wes测序数据(即生产数据样本比对后的wes bam文件),对两种数据进行分析,获得str相关疾病异常扩增的靶标区域数据(str变异及侧翼序列相关区域bam文件),并获得str变异及侧翼序列相关区域覆盖度等数据;
55.2)常规分析软件流程搭建和本地库构建:
56.例如以软件expansionhunter和exstra对不同样本的wes测序数据进行分析处理,分别构建本地阴性样本(即正常人)str样本集和本地特异基因str异常参考集;
57.3)软件预测和实验扩增次数差异评估
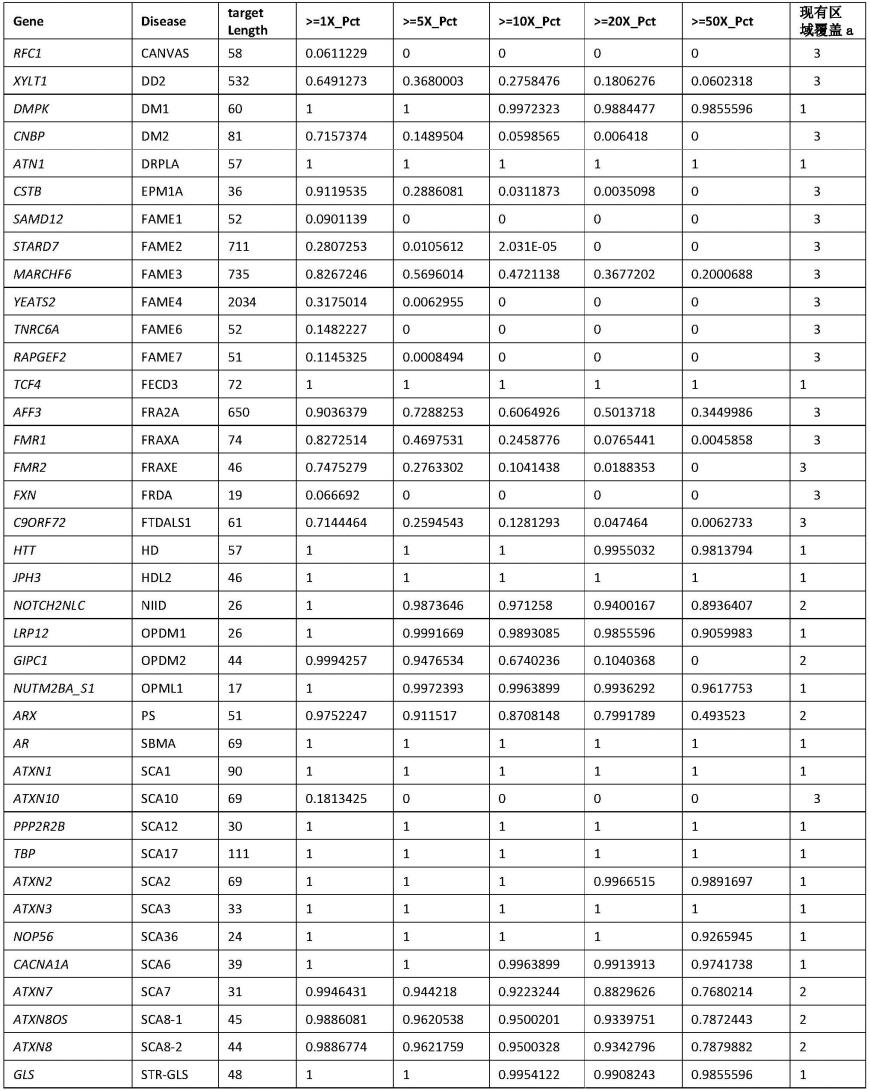
58.4)致病性str变异过滤指标和评估标准确定
59.根据str疾病相关异常过滤指标和评估标准对上述分析结果进行评估,将其列入重点关注的y标签或后续回顾的l标签。
60.6)疾病相关异常扩增str鉴定标准灵敏性和特异性评估。
61.本发明首先获取str相关疾病基因靶标区域内覆盖度情况,参见图2,图2为本发明实施例获取str相关疾病基因靶标区域内覆盖度情况的流程示意图。
62.本发明首先通过检索确定现有文献中公开的str相关疾病基因,例如使用关键词str、short tandem repeat或者genetic disorder等在omim数据库和pubmed等数据库中获取str相关疾病基因,共计38个基因,并确定疾病相关异常扩增的发生的基因组区域和疾病相关异常扩增范围,包括最小值min_abnorm和最大值max_abnorm。对于目标区域长度10bp以下的str区间,将区间上下游延伸至25bp,确定str相关疾病异常扩增的靶标区域数据,生成str-gene bed文件。
63.本技术从生产数据,即实际样本中获取wes测序数据(以bam文件保存),例如200个,统计str相关疾病异常扩增的靶标区域内所有碱基的覆盖度以及测序覆盖度达到1x、10x、20x等的碱基占比,以及预定覆盖度下碱基占比满足条件,例如大于95%的实际样本的比例。使用实际wes探针产品真实检测情况下的覆盖度而非产品bed文件的重叠值可以更好的确定靶标区域内有足够的reads覆盖用于评估异常扩增的情况。结果参见表1,表1为str相关疾病基因wes测序数据靶标区域内满足不同覆盖度样本占比。
64.表1 str相关疾病基因wes测序数据靶标区域内满足不同覆盖度样本占比
[0065][0066]
其中:现有区域覆盖a,1:完全;2:覆盖有波动;3:覆盖不好。
[0067]
本发明以expansionhunter和exstra作为常规分析软件,根据上述获取的38个疾病相关基因按照软件要求准备相关位点文件。使用软件的默认参数,对样本的wes测序数据进行分析,例如:
[0068]
expansionhunter:
[0069]
使用软件的默认参数,分析每个阳性样本,命令行如下:
[0070]
expansionhunter
‑‑
reads smp.bam
‑‑
reference reference.fa
‑‑
variant-catalog/path/hg38/variant_catalog.json
‑‑
output-prefix smp
[0071]
参数说明:
[0072]
‑‑
reads
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
待检测的bam文件
[0073]
‑‑
reference
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
参考基因组fasta文件
[0074]
‑‑
variant-catalog
ꢀꢀꢀ
已知变异信息的str位点文件
[0075]
‑‑
output-prefix
ꢀꢀꢀꢀꢀ
输出文件的前缀
[0076]
每个检测样本可获取候选位点预测的扩增次数。
[0077]
exstra:
[0078]
根据str-gene bed,对于每个str区域,分别向两侧各延伸500bp后获取新的str-gene flanking500 bed文件,并使用原始文件截取str-gene flanking500 bed中的区域bam,获取对照target bam文件。使用来自https://github.com/bahlolab/bio-str-exstra的perl脚本和模块来读取对照和检测样本target bam并生成str计数。使用r exstra包进行候选的str得分计算,包括p value、t值、差异可视化等。
[0079]
对约317个正常人样本(即阴性样本)的expansionhunter结果进行整理,统计本地正常人的str扩增次数,构建本地正常人数据库。提取阴性样本的扩增次数,排序后,选择low最小值和up次大值,使用这2个值作为正常人扩张次数的范围(norm_min_cg;norm_max_cg)。因为软件预测会存在一些假阳性,选择次大值是为了避免一些假阳性数据导致正常扩增范围上限增大而产生一些假阴性的数据。
[0080]
表2不同str基因本地正常人软件预测扩增次数
[0081][0082]
样本数据表示:检测样本中可提示该基因扩增次数数据的总样本数,对于覆盖度差或未覆盖的区域expansionhunter无法进行扩增次数的预测。
[0083]
本技术在建立本地库后,对软件预测结果和实际实验扩增次数的差异进行评估,从而对扩增次数数据进行修正。市场上成熟的动态突变检测产品常使用pcr+毛细管电泳的方式进行检测。常见的检测基因或疾病包括齿状核红核苍白球路易体萎缩症(drpla)、弗里德里希综合征(frda)、肯尼迪综合征(sbma)、脆x染色体综合征(fx)、强直性肌营养不良(dm)、亨廷顿舞蹈症(hd)、脊髓小脑共济失调八型(1-3型、6-8型、12型、17型)、脊髓小脑共济失调十型(1-3型、6-8型、12型、17型,frda,drpla)。汇总临床送检的动态突变产品相关的
实验检测扩增数据,并获取相关样本的wes数据,进行expansionhunter扩增次数预测,共计80个样本,涉及到13个基因,这些均为临床诊断率较高的基因。比较str疾病相关基因正常扩增范围内实际观测值和软件预测值的差异,结果参见图3,图3为str疾病相关基因正常扩增范围内实际观测值和软件预测值,图3中,x轴表示部分动态突变相关基因,y轴表示观察值-预测值的差值,蓝色表示两等位基因中最小值差,黄色表示两等位基因中最大值的差。结果发现观测值普遍小于预测值,而有少数几个基因观测值与预测值差异的波动范围较大,最高达20个重复的差异。比如sca17平均少20个重复,sca3平均少5个重复。sca2比较平均,都是2次重复的差异。表现最好的5个基因为atn1、atxn1、ppp2p2b、atxn2、htt、tbp其次为atxn3、cacna1a、atxn7、atxn8、dmpk。将每个str正常扩增范围内实际观察和软件预测的平均差值为diff,将具有diff的基因的对应的min_abnorm进行修订,min_abnorm修=min_abnorm-diff。
[0084]
在本发明的一些实施例中,采用候选致病性str变异过滤指标和评估标准对按照上文所述的方法获得的数据进行分类,参见图4,图4为候选致病性str变异过滤指标和评估标准流程示意图,具体过程如下:
[0085]
y标准过滤条件如下:这些记录需要根据患者临床症状进行重点关注。
[0086]
1)根据上文所述的方法获取相关数据,选择靶标区域》20x覆盖度样本占比大于90%的基因,满足条件基因作为l组起始入组基因。
[0087]
2)选择疾病相关最小异常扩增值在ngs测序读长范围内(即min_abnorm*扩增碱基单元数《150bp,满足条件基因作为y标准入组基因。
[0088]
3)选择检测样本对应基因最大扩增次数》norm_max_cg的数据;
[0089]
4)选择检测样本对应基因最大扩增次数》min_abnorm修的数据;
[0090]
5)选择exstra分析受检样本和对照库扩增差异显著性pvalue《0.05。
[0091]
l标准过滤条件如下:
[0092]
在满足y标准1)的条件下,选择满足3)、4)、5)任一标准的str扩增数据均作为l标准入组到后续大规模回顾性分析。
[0093]
本技术对38个str基因数据按照上述方法进行处理,获得结果如下:
[0094]
y标准入组基因为:atxn1、atxn2、atxn3、cacna1a、atxn7、ppp2r2b、tbp、dmpk、htt、atn1;
[0095]
l标准入组基因为:ppp2r2b、tbp、atxn1、atxn2、nop56、atxn3、cacna1a、atxn7、atxn8、jph3、htt、dmpk、ar、atn1、lrp12、tcf4、gls、notch2nlc、nutm2ba_s1。
[0096]
本技术还进一步对疾病相关异常扩增str鉴定标准灵敏性和特异性评估,具体如下:评估仅针对y标准入组基因(10个)进行,选择18个动态突变产品检测出疾病相关异常扩增基因样本进行wes测序,并按照上述流程进行分析,最终共获得的个具有与疾病相关异常扩增阳性的wes数据,共计180条记录,参见表3。其中符合y标准的17个,符合l标准48+17个。
[0097]
y标准下:
[0098]
灵敏度(tpr):true positive rate,描述识别出的所有正例占所有正例的比例;
[0099]
计算公式为:tpr=tp/(tp+fn)。tp:true positive,真阳性、fn:false negative,假阴性。
[0100]
tpr=17/17+1=94.4%;
[0101]
特异度(tnr):true negative rate,描述识别出的负例占所有负例的比例;
[0102]
计算公式为:tnr=tn/(fp+tn)。tn:true negative,真阴性,fp:false positive,假阳性
[0103]
tnr:(180-18)/(0+162)=100%
[0104]
即入组检测阳性样本中,y标准下,灵敏度为94.4%,特异度为100%。保证通过该方法标准为y的记录有足够的证据证明其为一个真的异常扩增。其中假阴性样本中出现的atxn3(sca3)因其exstra pvalue不满足要求而被定义为l标准。根据上文所述的实验和预测差异分析可见sca3的波动稍大,导致差异显著性统计出现偏差导致。l标准下的atxn3基因需要特别关注。
[0105]
表3阳性动态突变实验样本wes数据str检测结果
[0106][0107]
基于此,本发明提供了一种全外显子测序数据的处理方法,包括以下步骤:
[0108]
步骤s1、获取第一参考数据,所述第一参考数据包括参考样本的str相关疾病基因靶标区域内碱基覆盖度数据和预定的碱基覆盖度下的样本占比数据;
[0109]
获取第二参考数据,所述第二参考数据包括阴性参考样本扩增次数数据;
[0110]
步骤s2、获取检测样本数据,所述检测样本数据包括检测样本的str相关疾病基因靶标区域内碱基覆盖度数据、预定的碱基覆盖度下的样本占比数据和检测样本扩增次数数据;
[0111]
将所述检测样本的str相关疾病基因靶标区域内碱基覆盖度数据和预定的碱基覆
盖度下的样本占比数据与所述参考样本的str相关疾病基因靶标区域内碱基覆盖度数据和预定的碱基覆盖度下的样本占比数据进行比对,获得第一比对结果数据;
[0112]
若所述第一比对结果数据为不符合,则将所述检测样本扩增次数数据与所述阴性参考样本扩增次数数据进行比对,获得第二比对结果数据。
[0113]
本发明首先获取第一参考数据和第二参考数据,其过程具体如下:
[0114]
获取str相关疾病基因数据,确定str相关疾病异常扩增的靶标区域数据;
[0115]
获取参考样本的wes测序数据,对比所述参考样本的wes测序数据与所述str相关疾病异常扩增的靶标区域数据,获得参考样本的str相关疾病基因靶标区域内碱基覆盖度数据和预定的碱基覆盖度下的样本占比数据;
[0116]
采用expansionhunter软件对参考样本中阴性样本的wes测序数据进行分析,获得阴性参考样本扩增次数数据。
[0117]
本技术首先获取str相关疾病数据,确定str相关疾病异常扩增的靶标区域数据。在一个具体的实现方式中,本技术通过检索现有文献获取str相关疾病数据,例如在omim和pubmed等数据库中获取str相关疾病基因,并确定str相关疾病发生异常扩增的基因区域和范围,即靶标区域。具体而言,所述str相关疾病异常扩增的靶标区域数据包括基因片段长度最小值min_abnorm和基因片段长度最大值max_abnorm。为了提高后续数据处理的准确性,对于基因片段长度小于10bp的靶标区域,将其上下游序列进行延伸,例如延伸至25bp,获得str相关疾病异常扩增的靶标区域数据。
[0118]
本技术提供的处理方法包括获取参考样本的wes测序数据的步骤,在一些实施例中,所述参考样本的wes测序数据可以为检测机构获得的与str相关疾病相关的wes测序结果。统计str相关疾病异常扩增的靶标区域内所有碱基的覆盖度,确定参考样本的wes测序数据中str相关疾病异常扩增的靶标区域,并根据str相关疾病异常扩增的靶标区域内所有碱基的覆盖度确定参考样本的wes测序数据str相关疾病异常扩增的靶标区域中覆盖度达到1x、10x、20x、30x等的碱基占比,同时统计预定覆盖度下碱基占比满足条件,例如大于95%的参考样本的比例。
[0119]
本发明提供的处理方法包括获取第二参考数据的步骤,所述第二参考数据包括阴性参考样本扩增次数数据。在一些可能的实现方式中,本技术采用expansionhunter软件对参考样本中阴性样本的wes测序数据进行分析,获得阴性参考样本扩增次数数据,具体而言,所述阴性参考样本扩增次数数据为扩增次数范围,由扩增次数最小值(norm_min_cg)和扩增次数次大值(norm_max_cg)分别作为所述扩增次数范围的上限和下限。
[0120]
进一步的,所述第二参考数据还包括阳性参考样本扩增次数数据,其处理方法与阴性参考样本扩增次数数据获取方法类似,本技术在此不再赘述。
[0121]
在一些可能的实现方式中,所述第二参考数据还包括参考样本中阳性样本的str计算得分数据,具体而言,本技术采用exstra软件对所述参考样本的wes测序数据进行分析,获得所述参考样本中阳性样本的str计算得分数据,例如p值(p value)、t值、差异可视化等。
[0122]
在一些可能的实现方式中,本技术还可以包括对所述阴性参考样本扩增次数数据进行修正的过程,具体为:
[0123]
步骤1、获取阳性样本的实际扩增次数数据和wes测序数据;
[0124]
步骤2、采用expansionhunter软件对阳性样本的wes测序数据进行分析,获得阳性样本的预测扩增次数数据;
[0125]
步骤3、根据所述阳性样本的实际扩增次数数据和预测扩增次数,对所述阴性参考样本扩增次数数据进行修正。
[0126]
具体而言,步骤1、步骤2的过程与上文所述的获取过程相类似,本技术在此不再赘述。获得阳性样本的实际扩增次数和预测扩增次数后,计算两者的差值,并根据该差值对所述阴性参考样本扩增次数数据进行修正。
[0127]
本发明提供的处理方法还包括步骤s2:
[0128]
获取检测样本数据,所述检测样本数据包括检测样本的str相关疾病基因靶标区域内碱基覆盖度数据、预定的碱基覆盖度下的样本占比数据和检测样本扩增次数数据;
[0129]
将所述检测样本的str相关疾病基因靶标区域内碱基覆盖度数据和预定的碱基覆盖度下的样本占比数据与所述参考样本的str相关疾病基因靶标区域内碱基覆盖度数据和预定的碱基覆盖度下的样本占比数据进行比对,获得第一比对结果数据;
[0130]
若所述第一比对结果数据为不符合,则将所述检测样本扩增次数数据与所述阴性参考样本扩增次数数据进行比对,获得第二比对结果数据。
[0131]
所述步骤s2中获取检测样本数据的过程与步骤s1中获取第一参考数据和第二参考数据的过程类似,具体为:
[0132]
获取检测样本的wes测序数据,对比所述检测样本的wes测序数据与所述str相关疾病异常扩增的靶标区域数据,获得检测样本的str相关疾病基因靶标区域内碱基覆盖度数据和预定的碱基覆盖度下的样本占比数据;
[0133]
采用expansionhunter软件对检测样本的wes测序数据进行分析,获得检测样本扩增次数数据。
[0134]
获得检测样本数据后,将所述检测样本的str相关疾病基因靶标区域内碱基覆盖度数据和预定的碱基覆盖度下的样本占比数据与所述参考样本的str相关疾病基因靶标区域内碱基覆盖度数据和预定的碱基覆盖度下的样本占比数据进行比对,获得第一对比数据,例如预定的靶标区域的碱基覆盖度达到20x以上,碱基的样本占比数据为90%以上,若检测样本相应的数据满足上述预定条件,即将所述检测样品的数据列入l标准入组基因。
[0135]
若检测样本相应的数据不满足上述预定条件,则对其进行下一步判断。在一个具体的实施方案中,所述检测样本数据还包括检测样本异常扩增的发生的基因组区域和疾病相关异常扩增范围,包括最小值min_abnorm和最大值max_abnorm。如果检测样本相应的数据不满足上述预定条件时,继续比对所述检测样本的异常扩增的发生的基因组区域的最小值min_abnorm,获得第二比对结果数据。具体而言,将min_abnorm与ngs测序读长进行比对,若min_abnorm在ngs测序读长范围内,则将所述检测样本数据列入y标准入组基因,若min_abnorm不在ngs测序读长范围内,则对所述检测样本数据进行进一步的比对分析。
[0136]
具体而言,进一步比对分析包括:将检测样本数据中的最大扩增次数与所述阴性样本的扩增次数数据进行比对分析,包括以下比对分析的至少一种:
[0137]
(1)将所述检测样本数据的最大扩增次数与所述阴性样本的扩增次数的次大值(norm-max-cg)进行比对分析,如该最大扩增次数不大于norm-max-cg,则将该数据列为l标准入组基因,如该最大扩增次数大于norm-max-cg,则列为y标准,作为临床解读重点关注数
据;
[0138]
(2)将所述检测样本数据的最大扩增次数与所述修正后的阴性样本的扩增次数的最小值(min_abnorm修)进行比对分析,若该最大扩增次数不大于修正后的阴性样本的扩增次数的最小值,则将该数据列为l标准入组基因,若该最大扩增次数大于修正后的阴性样本的扩增次数的最小值,则列为y标准,作为临床解读重点关注数据;
[0139]
(3)获取所述检测样本数据中采用exstra计算获得的p值,若该p值≥0.05,则将数据列为l标准入组基因,若p值《0.05,则列为y标准,作为临床解读重点关注数据。
[0140]
本发明提供了一种全外显子测序数据的处理系统,包括第一参考数据获取单元,所述第一参考数据单元用于获取第一参考数据,所述第一参考数据包括参考样本的str相关疾病基因靶标区域内碱基覆盖度数据和预定的碱基覆盖度下的样本占比数据;
[0141]
第二参考数据获取单元,所以第二参考数据获取单元用于获取第二参考数据,所述第二参考数据包括阴性参考样本扩增次数数据;
[0142]
检测样本数据获取单元,所述检测样本数据获取单元用于获取检测样本数据,所述检测样本数据包括检测样本的str相关疾病基因靶标区域内碱基覆盖度数据、预定的碱基覆盖度下的样本占比数据和检测样本扩增次数数据;
[0143]
第一比对单元,所述第一比对单元用于将所述检测样本的str相关疾病基因靶标区域内碱基覆盖度数据和预定的碱基覆盖度下的样本占比数据与所述参考样本的str相关疾病基因靶标区域内碱基覆盖度数据和预定的碱基覆盖度下的样本占比数据进行比对,获得第一比对结果数据;
[0144]
第二比对单元,所述第二比对单元用于比对所述检测样本扩增次数数据与所述阴性参考样本扩增次数数据,获得第二比对结果数据。
[0145]
在一个具体的实现方式中,所述第一参考数据获取单元包括str相关疾病基因数据获取单元,所述str相关疾病基因数据获取单元用于获取str相关疾病基因数据,确定str相关疾病异常扩增的靶标区域数据;
[0146]
参考样本wes测序数据获取单元,所述参考样本wes测序数据获取单元用于获取参考样本的wes测序数据;
[0147]
第三比对单元,所述第三比对单元用于对比所述参考样本的wes测序数据与所述str相关疾病异常扩增的靶标区域数据,获得参考样本的str相关疾病基因靶标区域内碱基覆盖度数据和预定的碱基覆盖度下的样本占比数据。
[0148]
在一个具体的实现方式中,所述检测样本数据获取单元包括检测样本wes测序数据获取单元,所述检测样本wes测序数据获取单元用于获取检测样本的wes测序数据;
[0149]
第四比对单元,所述第四比对单元用于对比所述检测样本的wes测序数据与所述str相关疾病异常扩增的靶标区域数据,获得检测样本的str相关疾病基因靶标区域内碱基覆盖度数据和预定的碱基覆盖度下的样本占比数据;
[0150]
检测样本扩增次数数据处理单元,用于采用min_abnorm软件对检测样本的wes测序数据进行分析,获得检测样本扩增次数数据。
[0151]
本发明提供的数据处理系统用于实现上述数据处理方法,各单元的作用在于实现数据处理方法的各步骤,本技术在此不再赘述。
[0152]
本发明还提供了一种检测短串联重复疾病相关异常扩增的系统,包括第一参考数
据获取单元,所述第一参考数据单元用于获取第一参考数据,所述第一参考数据包括参考样本的str相关疾病基因靶标区域内碱基覆盖度数据和预定的碱基覆盖度下的样本占比数据;
[0153]
第二参考数据获取单元,所以第二参考数据获取单元用于获取第二参考数据,所述第二参考数据包括阴性参考样本扩增次数数据;
[0154]
检测样本数据获取单元,所述检测样本数据获取单元用于获取检测样本数据,所述检测样本数据包括检测样本的str相关疾病基因靶标区域内碱基覆盖度数据、预定的碱基覆盖度下的样本占比数据和检测样本扩增次数数据;
[0155]
第一比对单元,所述第一比对单元用于将所述检测样本的str相关疾病基因靶标区域内碱基覆盖度数据和预定的碱基覆盖度下的样本占比数据与所述参考样本的str相关疾病基因靶标区域内碱基覆盖度数据和预定的碱基覆盖度下的样本占比数据进行比对,获得第一比对结果数据;
[0156]
第二比对单元,所述第二比对单元用于比对所述检测样本扩增次数数据与所述阴性参考样本扩增次数数据,获得第二比对结果数据;
[0157]
预测系统,所述预测系统用于根据第一比对结果数据和第二比对结果数据,获得短串联重复疾病相关异常扩增的预测结果。
[0158]
具体而言,所述预测系统用于对所述第一比对结果数据和第二比对结果数据进行分析,获得短串联重复疾病相关异常扩增的预测结果。获得检测样本数据后,将所述检测样本的str相关疾病基因靶标区域内碱基覆盖度数据和预定的碱基覆盖度下的样本占比数据与所述参考样本的str相关疾病基因靶标区域内碱基覆盖度数据和预定的碱基覆盖度下的样本占比数据进行比对,获得第一对比数据,例如预定的靶标区域的碱基覆盖度达到20x以上,碱基的样本占比数据为90%以上,预测系统根据所述第一对比数据判断将该检测样本列入l入组标准基因还是进行下一步判断,若检测样本相应的数据满足上述预定条件,即将所述检测样品的数据列入l标准入组基因。
[0159]
若检测样本相应的数据不满足上述预定条件,则对其进行下一步判断。在一个具体的实施方案中,所述检测样本数据还包括检测样本异常扩增的发生的基因组区域和疾病相关异常扩增范围,包括最小值min_abnorm和最大值max_abnorm。如果检测样本相应的数据不满足上述预定条件时,继续比对所述检测样本的异常扩增的发生的基因组区域的最小值min_abnorm,获得第二比对结果数据。具体而言,将min_abnorm与ngs测序读长进行比对,若min_abnorm在ngs测序读长范围内,预测系统将所述检测样本数据列入y标准入组基因,若min_abnorm不在ngs测序读长范围内,,则对所述检测样本数据进行进一步的比对分析。
[0160]
具体而言,进一步比对分析包括:将检测样本数据中的最大扩增次数与所述阴性样本的扩增次数数据进行比对分析,包括以下比对分析的至少一种:
[0161]
(1)将所述检测样本数据的最大扩增次数与所述阴性样本的扩增次数的次大值(norm_max_cg)进行比对分析,如该最大扩增次数不大于norm_max_cg,则将该数据列为l标准入组基因,如该最大扩增次数大于norm_max_cg,则列为y标准,作为临床解读重点关注数据;
[0162]
(2)将所述检测样本数据的最大扩增次数与所述修正后的阴性样本的扩增次数的最小值(min_abnorm修)进行比对分析,若该最大扩增次数不大于修正后的阴性样本的扩增
次数的最小值,则将该数据列为l标准入组基因,若该最大扩增次数大于修正后的阴性样本的扩增次数的最小值,则列为y标准,作为临床解读重点关注数据;
[0163]
(3)获取所述检测样本数据中采用exstra计算获得的p值,若该p值≥0.05,则将数据列为l标准入组基因,若p值《0.05,则列为y标准,作为临床解读重点关注数据。
[0164]
本发明通过wes测序数据中实际样本真实覆盖度情况定义样本可检测的str相关基因,比使用wes探针bed区域/bed+flanking区域的是否重叠来评估更准确。本发明提供的全外显子测序数据的处理方法受不同的算法、不同的测序平台、不同的探针、不同的比对软件影响较小,得到的数据结果较为准确。
[0165]
以上仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1