一种分析肠道菌群中致病菌和量化菌群地域特征的方法与流程
1.本发明属于人体肠道菌群检测领域,本发明涉及利用生物信息学技术对与人体健康相关的数百种致病菌进行高通量、高精度检测。并且能够量化描述肠道菌群的种族和地域特征。
背景技术:
2.肠道菌群就像是人体的另外一个器官,与我们的健康息息相关。人体肠道内居住着成千上万的细菌,而每个人肠道中的菌群特征是不同的。菌群特征的差异性又可以进一步关联到不同个体的生理特征或是健康状态。比如,通过检测肠道菌群中的致病菌种类,我们直接推断出某个体可能会面临的感染风险。世界卫生组织/国际癌症研究机构将幽门螺旋杆菌定为i类致癌原,因此如果某个体的肠道菌群中积累了足够量的幽门螺旋杆菌,我们可以及时预警患肠胃癌的风险。除了检测像致病菌这类与人体健康直接相关的微生物,肠道菌群还带有显著的地域特征。大量的研究表明,来自不同种族和地域的人群呈现出不同的菌群特征。因此,我们可以利用肠道菌群数据来量化菌群特征与个体种族或地域特征之间的吻合程度。这不仅能帮助我们更好的了解每个人肠道的特点,而且有利于提升基于肠道菌群的诊断和治疗方法的有效性。
3.目前针对致病菌的检测,常见的手段是通过采集人体组织样本(比如,血液、粪便、皮肤组织等),然后在实验室进行培养、观察、pcr扩增特异性引物,最后由专业人士鉴定。这种检测方式明显不足之处包括:1)致病菌检测范围小;2)需要一定的人力、时间和经济成本;3)容易引入实验误差和专业人员的主观判断偏差。另外,随着测序技术的快速发展,下一代测序技术开始成为一种重要的检测工具。其中,对单个菌株进行全基因组水平测序能够捕捉到完整的该菌株的基因组信息,但是在检测大量菌种场景中,由于价格高昂,很难应用于实际生产中。16s rrna基因测序在成本上有明显优势,能够同时对肠道中大量细菌进行检测,但是目前大部分基于16s rrna基因的宏基因组分析技术仅能鉴定到“属”级以上,而不能对差异性更大的“种”级进行鉴定。
4.最近,一些方法对基于16s rrna宏基因组分析方法进行了优化
[1-2]
,他们能够实现仅依赖于16s rrna也能将菌群中大部分的微生物鉴定到“种”级别。但是他们都缺乏对致病菌数据的细致整理(比如,详细致病性的的介绍、常见感染源、和相关预防建议等),同时也缺少对肠道菌群族和地域域特征分析的功能。这些功能不仅在辅助诊断领域起着重要作用,而且大大扩展了肠道菌群数据的商业应用场景。本发明的目的在提升利用16s rrna全长测序信息在肠道菌群“种”水平的检测能力,同时扩展致病菌的检测范围和丰富致病菌的相关信息(比如,致病菌的来源,致病性,常见感染症状和科学预防建议)和量化待分析样本菌群构成与不同地区人群菌群构成的相似性。
技术实现要素:
[0005]
为解决上述技术问题,本发明基于16s rrna基因全长(v1-v9区间)测序信息,通过
生物信息学技术与搭载在本发明中的数万种肠道菌群的16s rrna基因数据库进行对比,从而检测出所测样本中肠道菌群的种类和载量。
[0006]
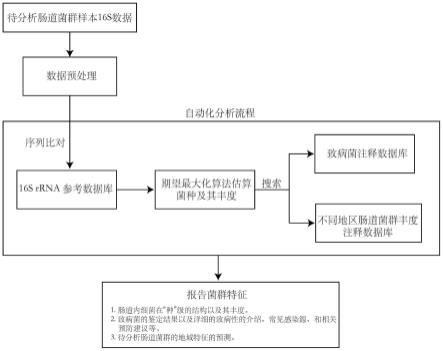
本发明第一方面,公开了一种基于期望最大化算法的自动化分析流程,其用来鉴定肠道菌群中的微生物种类和量化其在样本中的丰度,其操作流程如图1所示。
[0007]
具体操作流程为:
[0008]
1.1)对输入的16s rrna数据进行预处理,包括:
[0009]
(1)去除低质量测序数据;
[0010]
(2)只保留完整或者趋于完整的16s rrna测序片段(长度在1200-1600bp);
[0011]
(3)将由测序仪生成的fastq文件格式转换为下游分析所需要的fasta文件;
[0012]
优选的,步骤(1)中,低质量测试数据为nanopore测序所得数据中平均片段质量低于9的测序片段;
[0013]
优选的,步骤(2)中,完整或者趋于完整的16s rrna测序片段长度在1200-1600bp;
[0014]
1.2)加载16s rrna全长基因参考数据库到分析流程,分析流程根据数据库所包含的细菌种类,为所有细菌设置相同的初始丰度(比如,数据库包含n种细菌,则每种细菌的丰度为1/n);
[0015]
1.3)分析流程调用序列比对软件(比如,minimap2,bowtie2或者bwa)
[3-5]
,将待分析样本中的每条16s rrna片段与参考数据库中的16s rrna片段进行一对一比对。分析流程将每对序列之间各个位点的比对结果记录下来,然后计算序列吻合概率。两条序列之间每对核酸的比对结果通过不同位点比对类型来表达。所有位点类型包括:错配位点(x),缺失或者插入位点(indel),剪切位点(s)和吻合位点(m);
[0016]
1.4)通过每对序列之间不同的位点比对类型结果来计算两条序列吻合的概率p(a),c来自于所有位点比对类型c=[x,indel,s,m],p(c)为某个位点比类型的概率(比如,错配位点(x)的概率为p(x)=nx/n,nx为x类位点的个数,n为所有位点类型的总和)。当待分析样本中的多个16s rrna片段与数据库中同一个参考16s rrna序列吻合时,分析流程记录下所有吻合概率中的最大值。因为参考数据库中的每一条16s rrna基因对应一种细菌,所以该细菌在待分析样本中检测到的概率即等于待分析16s rrna基因片段与参考16s rrna基因片段之间的概率p(a)。
[0017]
1.5)将步骤1.2)中生成的所有细菌的初始丰度与步骤1.4)中所得的该细菌的概率导入期望最大化模型中迭代优化。期望最大化模型会基于上一次的分析结果来调整各种细菌的丰度以增加整体估算概率,经过多次迭代优化后,整体估算概率达到最大值,迭代终止。去除低概率菌种过后,分析所得的菌种类型及其丰度既可以反应待分析样本中菌群的构成和相应的丰度。
[0018]
本发明第二方面,公开了一种高质量16s rrna全长基因参考数据库。
[0019]
所述数据库中,标记每条16s rrna基因序列标注完整的细菌分类学命名(包括界、门、纲、目、科、属、种)。通过文献梳理,将参考数据库中的致病菌进行标注整理,生成一个关于包含致病菌详细信息的数据库。
[0020]
优选的,所述致病菌详细信息包括致病菌的来源,致病性,常见感染症状和科学预
防建议。
[0021]
优选的,数据库中16s rrna 来源于原核生物;
[0022]
进一步优选的,去除掉完全相同的16s rrna序列和具有不同分类学名称但是序列完全相同的16s rrna序列;
[0023]
优选的,原核生物16s rrna参考序列来源包括但不限于下表1所列物种或数据库来源:
[0024]
表1常见致病菌物种
[0025]
[0026]
[0027][0028]
distance)、杰卡德距离(jaccard distance)等机器学习和统计分析方法。
[0046]
本发明和现有技术相比,具有以下有益效果:
[0047]
(1)本方案中新开发的期望最大化算法能够有效利用完整16s rrna基因的信息来达到在分类学水平鉴定出更多菌种的效果。得益于期望最大化算法多次迭代优化的功能,一些实验操作过程或者测序过程中引入的误差能够在迭代优化过程中过滤掉,最后有效地提高了检测精度。
[0048]
(2)本方案中的自建数据库通过大量文献搜索和人工校正,保证了参考数据的高质量,并且数据库中每一条16s rrna都是独特唯一且注释了正确的菌种分类学命名。因此,所有检测到的菌种都有对应的菌种分类学名称(包括界、门、科、目、冈、属、种)。
[0049]
(3)本方案中人工整理的致病菌数据库,不仅在致病菌种类上明显高于公共开放数据库(https://www.ncbi.nlm.nih.gov/pathogens/),而且我们还通过海量文献搜索整理,将各个致病菌的来源,致病性,常见感染症状和科学预防建议等信息收录在内。这样,当使用本方案检测致病菌时,尽管无专业人士辅助解读结果,相关检测结果也一目了然。
[0050]
(4)利用具有地理位置信息的宏基组测序数据来构建含有地理位置信息的菌群丰度数据库,当新测的16s rrna样本通过本方法进行菌种鉴定后,再与该丰度数据库进行比对,从而得出该样本与不同地区人群的肠道菌群相似度。因此,丰富了对未知肠道菌群数据的描述方法。
附图说明
[0051]
图1为本发明整体流程示意图。
[0052]
图2为本发明与mapseq在微生物分类学各个水平的检测能力的差异。
具体实施方式
[0053]
下面将结合示意图对本发明公开内容进行更详细的描述,其中表示了本发明的优选实施例,应该理解本领域技术人员可以修改在此描述的本发明,而仍然实现本发明的有利效果。因此,下列描述应当被理解为对于本领域技术人员的广泛知道,而并不作为对本发明的限制。
[0054]
实施例1,一种分析肠道菌群中致病菌和量化菌群地域特征的方法,其操作流程如图1所示。
[0055]
具体包括以下步骤:
[0056]
步骤1)构建一种基于期望最大化算法的自动化分析流程,用来快速精准地鉴定待分析样本中的微生物种类和对应的丰度
[0057]
1.1)分析流程首先对输入的16s rrna数据进行预处理:
[0058]
(1)去除低质量测序数据(比如,nanopore测序所得数据中平均片段质量低于9的测序片段为质量过低);
[0059]
(2)只保留完整或者趋于完整的16s rrna测序片段(长度在1200-1600bp);
[0060]
(3)将由测序仪生成的fastq文件格式转换为下游分析所需要的fasta文件;
[0061]
1.2)分析流程紧接着根据数据库所包含的细菌种类的总数,为所有细菌生成相同的初始丰度(比如,数据库包含n种细菌,则每种细菌的丰度为1/n);
[0062]
1.3)分析流程通过序列比对软件(比如,minimap2,bowtie2或者bwa)
[3-5]
,将待分析样本中的每条16s rrna片段与所选择的参考数据库中的16s rrna 片段进行一对一比对。然后,分析流程将每对序列之间各个位点的比对结果记录下来,用来计算序列吻合概率。两条序列之间每对核酸的比对结果通过不同位点比对类型来表达。所有位点类型包括:错配位点(x),缺失或者插入位点(indel),剪切位点(s)和吻合位点(m);
[0063]
1.4)利用每对序列之间不同的位点比对类型结果来计算两条序列吻合的概率p(a),c来自于所有位点比对类型c=[x,indel,s,m],p(c)为某个位点比类型的概率(比如,错配位点(x)的概率为p(x)=nx/n,nx为x类位点的个数,n为所有位点类型的总和)。当待分析样本中有多个16srrna片段与参考数据库中同一个16srrna序列吻合时,分析流程记录下所有吻合概率中的最大值。因为参考数据库中的每一条16srrna基因对应一种细菌,所以该细菌在待分析样本中检测到的概率即等于待分析16srrna基因片段与参考16srrna基因片段之间的吻合概率p(a)。
[0064]
1.5)将步骤1.2)中生成的所有细菌的初始丰度与步骤1.4)中所得的该细菌的概率导入期望最大化模型中迭代优化。期望最大化模型基于上一次的分析结果来调整各种细菌的丰度以增加整体估算概率,经过多次迭代优化后,整体估算概率达到最大值,迭代终止。去除低概率菌种过后,分析所得的菌种类型及其丰度既可以反应待分析样本中菌群的构成和相应的丰度。
[0065]
步骤2)构建高质量的16s rrna参考数据库,并且注释完整的致病菌子数据库:
[0066]
2.1)获取原核生物16s rrna参考序列;
[0067]
2.2)去除掉长度不在1400bp-1600bp范围内的16s rrna序列、去除掉完全相同的16s rrna序列和具有不同分类学名称但是序列完全相同的16s rrna序列、去除掉没有精确到微生物种级分类学命名的16s rrna序列;
[0068]
2.3)通过搜集文献,将参考数据库中的致病菌进行标注,并且建立一个对标注致病菌有详细描述的子数据库,包括信息:致病菌的来源,致病性,常见感染症状和科学预防建议。
[0069]
说明:16s rrna基因参考数据库和致病菌注释数据库可以分别根据2.2)和2.3)所描述方式进行扩展作为本发明所述步骤2.1)原核生物16s rrna参考序列来源包括但不限于:
[0070]
https://rrndb.umms.med.umich.edu/;
[0071]
https://www.ncbi.nlm.nih.gov/bioproject/?term=prjna33175;
[0072]
https://www.ncbi.nlm.nih.gov/bioproject/prjna33317。
[0073]
步骤3)构建具有地理位置特征的菌群丰度数据库:
[0074]
3.1)获取不同国家或地区的人体肠道宏基因组数据;
[0075]
3.2)将3.1)所获得的人体肠道宏基因组数据进行预处理:(1)将测序读长数量小于5000000的样本去掉;(2)将没有明确地理位置的样本去掉;
[0076]
3.3)利用metaphlan2
[6]
对每个宏基因组样本中的菌群结构进行量化,计算出每个菌种在样本中的丰度;
[0077]
3.4)建立不同地区的肠道菌群丰度注释数据库。
[0078]
作为本发明所述步骤3.1)不同国家或地区的人体肠道宏基因组数据来源包括但不限于:来自于https://www.ncbi.nlm.nih.gov/的53个宏基因组项目样本(具体样本编号请参考表1)
[0079]
说明:数据库可以根据最新研究进展和按照3.2)和3.3)的构建方法进行扩容,也可以添加自行测序数据。
[0080]
步骤4)准备待分析样本的16s rrna数据并且导入分析流程进行处理:
[0081]
4.1)粪便样本采集后立即保存于dna保存液中,然后利用粪便微生物dna提取试剂盒将保存液中的粪便微生物dna提取出来。提取后的dna在纯化和稀释过后,用特定的pcr引物来扩增16s rrna基因全长片段。将扩增后的产物在下一代测序仪上进行测序,即得到所述待分析粪便样本16s rrna的测序片段;
[0082]
4.2)将所述4.1)中产生的16s rrna测序结果导入所述步骤1)已构建好的自动化分析流程中,得到该样本中所包含的细菌种类以及其丰度。将所检测到的细菌种类在所述步骤2)中的致病菌数据库中检索,从而获得所检测到的细菌中的致病菌种类以及关于所检测到的致病菌的详细说明与预防建议;同时,将所检测到的细菌种类与步骤3)所述的已构建好的具有地理位置特征的菌群丰度数据库进行对比,量化待分析样本菌群与不同地区人群菌群的相似度。相似度的量化手段可以采用包括但不限于随机森林(random forest)、布雷-柯蒂斯相异度(bray-curtis dissimilarity)、欧几里得距离(euclidean distance)、杰卡德距离(jaccard distance)等机器学习和统计分析方法。
[0083]
实施例2,从新样本中测序获得的16s rrna序列进行肠道菌群“种”水平分析
[0084]
具体步骤包括:
[0085]
利用粪便微生物dna提取试剂盒富集和纯化待分析粪便样本中的微生物dna,然后使用特定的pcr引物从提取出的dna中扩增获得16s rrna基因全场或接近全长的片段,最后通过下一代测序技术获得待分析粪便样本中16s rrna的测序数据。
[0086]
将步骤1)中获得的粪便样本16s rrna测序数据输入已构建好的如实施例1中所描述的自动化分析流程,获得该样本的肠道菌群结构分析结果,包括该样本所含有的细菌种类、致病菌种类、来源、致病性、常见症状和有效预防措施、以及该样本与不同地区人群肠道菌群的相似性。
[0087]
我们对12个中国人的28个粪便样本进行采样分析(其中包括来自同一个体的多个样本)。首先采用步骤1)中所描述的方式获得样本中的16s rrna基因测序数据,每个样本最低测序深度为2.5万序列。首先,为了评估该发明在菌种鉴定能力方面的效果,我们用本发明和另一个在本领域广泛使用的方法mapseq v1.2.6
[7]
分别对本实施例子中的新测数据进行分析。
[0088]
作为比较结果,从图2中可以看出,从分类学水平“科”以下(“科”,“属”和“种”),在菌种的检测能力方面本发明明显优于本领常用软件(mapseq)。特别在“属”和“种”水平,本发明的检测能力分别提升了约3倍和6倍左右(平均值)。
[0089]
从表3中可以获得单个样本中所检测到的致病菌种类,另外关于每种所检测出的致病菌详细的描述和对应改善建议(基于学术文献)也由本方法自动生成。
[0090]
表3.单个样本举例,检测出的致病菌结果展示。
sequences.”bioinformatics 34(18):3094
–
3100.
[0100]
[5]li,heng,and richard durbin.2009.“fast and accurate short read alignment with burrows-wheeler transform.”bioinformatics 25(14):1754
–
60.
[0101]
[6]truong,duy tin,eric a.franzosa,timothy l.tickle,matthias scholz,george weingart,edoardo pasolli,adrian tett,curtis huttenhower,and nicola segata.2015.“metaphlan2 for enhanced metagenomic taxonomic profiling.”nature methods 12(10):902
–
3.
[0102]
[7]matias rodrigues,f.,thomas s.b.schmidt,janko tackmann,and christian von mering.2017.“mapseq:highly efficient k-mer search with confidence estimates,for rrna sequence analysis.”bioinformatics 33(23):3808
–
10.
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1