一种FL118或其7位结构修饰衍生物的药物组合物以及制备方法和应用
一种fl118或其7位结构修饰衍生物的药物组合物以及制备方法和应用
技术领域
1.本发明涉及一种fl118或其7位结构修饰衍生物的药物组合物以及制备方法和应用。
背景技术:
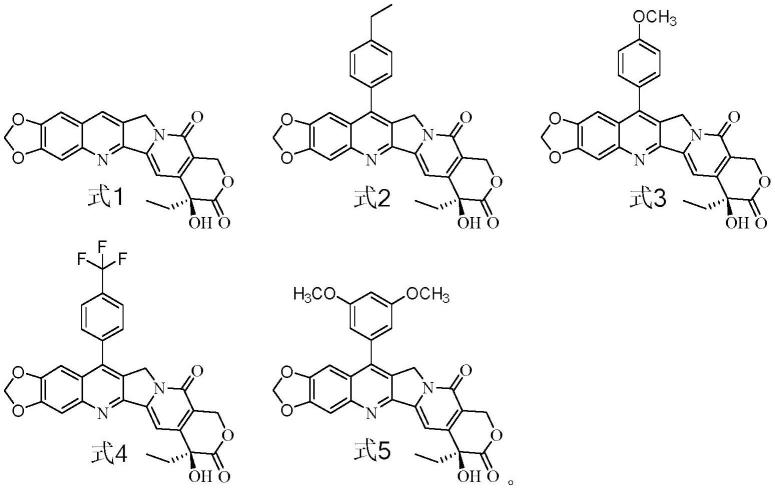
2.fl118(式1)(10,11-methylenedioxy-20s-camptothecin)是一种全合成化合物,是美国研究者利用survivin高通量筛选技术发现的一种小分子抗癌药,前期研究已发现fl118对于人头颈癌(fadu)、人结肠癌(sw620)和人肺癌(ekvx)等均有良好的抑制作用,能作用于产生耐药机制的多个靶点,能抑制多种抗凋亡蛋白;对多种耐药的肿瘤细胞的抑制效果明显高于其他抗癌药物,体内动物试验中的抗肿瘤效果也优于目前的临床药物,且在动物体内也体现较弱毒性,是非常有前景的候选化合物,尽管fl118有上述活性优势,但其同时具有难溶性和低渗透性的不利于成药的特征。dr li fengzhi等学者曾采用环糊精、dmso、peg混合液提升fl118溶解度(li fengzhi,ling xiang,cao shousong,novel formulations of water-insoluble chemical compounds and methods of using a formulation of compound fl118 for cancer therapy,wo2012/058666a2,国际申请号:pct/us2011/058558,优先日:2010.10.29),但始终未能实现其高溶解性和高渗透性的成功改进。
3.化合物7-(4-乙基苯基)-fl118(式2)、7-(4-甲氧基苯基)-fl118(式3)、7-(4-三氟甲基苯基)-fl118(式4)和7-(3,5-二甲氧基苯基)-fl118(式5)是以式1(fl118)为母核经过结构修饰得到的衍生物,申请人曾尝试用dr li fengzhi专利报到的方法改善式4化合物的溶解度和生物利用度(王孟可,洪亦超,夏利华,冯亚男,纪东浩,王文超,李庆勇,7-p-tfm-fl118-β-环糊精包合物的药动学和组织分布研究,中国药学杂志2019年4月第54卷第7期576-580),溶解度虽然有所提升,但对靶器官细胞的吸收利用没有明显改进,该处方中的有机溶剂用量仍是限制使用的瓶颈。
4.具有显著抗肿瘤活性的化合物式1~式5虽然结构式已经公开多年,但其难溶性和低渗透性的特征无法实现靶器官药物的药效浓度,严重阻碍此类化合物进一步开发利用。因此本发明公开的一种式1~式5结构类型化合物的药物组合物,该组合物组成避开了有机溶剂,将此类化合物溶解度、释放度、靶器官吸收度同时提升到满足有效药用范围,同时该组合物具备制药过程可接受的稳定性,解决此类化合物成药过程的关键技术问题。
技术实现要素:
5.fl118及其7位结构修饰衍生物很久以来因为难溶性、低渗透性和低靶细胞的吸收利用度未能制备成适当的剂型用于动物实验,在以往动物实验中,研究者通过提高给药剂量来实现靶器官有效药物浓度所造成的毒副作用非常明显,从而限制了此类化合物进一步临床开发。本发明制备的此类化合物的药物组合物能够实现:
6.1)显著提升此类化合物在水中的溶解度和释放度,充分满足此类化合物有效药用浓度1-5mg/ml的药用需求;
7.2)本发明制备的药物组合物显著提高了靶细胞的吸收利用度,使在低给药剂量下实现靶细胞的药效浓度,降低毒副作用风险,显著优于已有报道技术;
8.3)本发明所公开的药物组合物组方和制备方法能提供任何比例的制药所需要浓度,组合物的稳定性满足制药工艺需求,可以作为现成的固体粉末直接给药,也可以添加少量其他辅料做成口服剂型给药。
9.本发明的技术方案如下:
10.一种药物组合物,由活性成分fl118或其7位结构修饰衍生物、soluplus(聚乙烯己内酰胺-聚醋酸乙烯酯-聚乙二醇接枝共聚物)、羟丙基甲基纤维素邻苯二甲酸酯组成;
11.其中,活性成分与soluplus的质量比为1:5~1:40,活性成分与soluplus的质量之和与羟丙基甲基纤维素邻苯二甲酸酯的质量比为100:1~1:5;
12.优选的,活性成分与soluplus的质量比为1:12~1:20,活性分子与soluplus的质量之和与羟丙基甲基纤维素邻苯二甲酸酯的质量比为20:1~10:1;
13.所述活性成分fl118如式1所示,fl118的7位结构修饰衍生物如式2、式3、式4或式5所示:
[0014][0015]
本发明药物组合物的制备方法可以是旋转蒸发法、喷雾干燥法中的任意一种。具体的制备方法如下:
[0016]
将活性成分与soluplus溶解在溶剂中,经旋转蒸发或喷雾干燥去除溶剂,得到无定型药物组合物前体粉末,将该前体粉末与羟丙基甲基纤维素邻苯二甲酸酯物理混合,即得本发明药物组合物;
[0017]
所述溶剂选自丙酮、乙腈、二氯甲烷、三氯甲烷、甲醇、乙醇、四氢呋喃、异丙醇中的一种或多种;
[0018]
所述旋转蒸发去除溶剂的温度为25~55℃,旋转蒸发的时间应大于10min;
[0019]
所述喷雾干燥的参数为:入口温度55~85℃、出口温度≥60℃、雾化气流量335~
415l/h、蠕动泵速度2~5ml/min。
[0020]
本发明所述的药物组合物可进一步与适宜的药用辅料进行处理,制成片剂、胶囊剂、颗粒剂、散剂或混悬剂等口服制剂。
[0021]
本发明所述的药物组合物可应用于制备抗癌药物,尤其是在制备治疗肺癌、膀胱癌、乳腺癌、宫颈癌、卵巢癌、胰腺癌、小肠癌、胃癌、肝癌、结肠癌和头颈癌的药物中的应用。
[0022]
本发明的有益效果在于:
[0023]
本发明公开了一种fl118或其7位结构修饰衍生物的药物组合物及制备方法和应用,该药物组合物中包含fl118或其7位结构修饰衍生物作为活性成分,辅料包含聚乙烯己内酰胺-聚醋酸乙烯酯-聚乙二醇接枝共聚物soluplus和羟丙基甲基纤维素邻苯二甲酸酯。采用旋转蒸发、喷雾干燥法中的任意一种方法制备获得fl118或其7位结构修饰衍生物的药物组合物。
[0024]
所述的药物组合物中fl118或其7位结构修饰衍生物活性成分以无定型态存在,显著提升此类化合物在水中的溶解度和释放度,显著提升靶细胞的吸收利用度,满足此类化合物有效药用浓度1-5mg/ml的药用需求。
[0025]
本发明药物组合物的组方和制备方法能提供任何比例的制药所需要浓度,组合物的稳定性满足制药工艺需求,可以作为现成的固体粉末直接给药,也可以添加少量其他辅料做成口服剂型给药,应用于肿瘤治疗。
附图说明
[0026]
图1为本发明实施例1,2,3,4,5,6中化合物4无定型药物组合物前体粉末在蒸馏水中的溶出释放曲线。
[0027]
图2为本发明实施例5中化合物4无定型药物组合物前体粉末以及其原料、载体、物理混合物的xrd扫描图。
[0028]
图3为本发明实施例5中化合物4无定型药物组合物前体粉末以及其原料、载体、物理混合物的dsc扫描图。
[0029]
图4为本发明实施例5中化合物4无定型药物组合物前体粉末、环糊精处理的化合物4、有机溶剂常规配制化合物4在靶细胞上的吸收摄取实验。
[0030]
图5为本发明实施例5和实施例6中化合物4无定型药物组合物前体粉末、物理混合物、环糊精制剂在雌性sd大鼠体内的药时曲线。
[0031]
图6为本发明实施例1和实施例8在室温存放6个月后的溶出释放曲线。
[0032]
图7为本发明实施例2和实施例9在室温存放6个月后的溶出释放曲线。
[0033]
图8为本发明实施例5和实施例10在室温存放6个月后的溶出释放曲线。
[0034]
图9为本发明实施例6和实施例11在室温存放6个月后的溶出释放曲线。
具体实施方式
[0035]
下面通过具体实施例进一步描述本发明,但本发明的保护范围并不仅限于此。
[0036]
实施例1
[0037]
按一定重量比例(1:20)分别称取活性小分子(式1-式5)与soluplus混合均匀,向混合物中加入二氯甲烷,在常温下充分震荡澄清后,通过旋转蒸发仪40度下去除溶剂,得到
式1-式5化合物的无定型药物组合物前体粉末。
[0038]
活性小分子(式1-式5)与soluplus物理混合物的制备:按一定重量比例(1:20)分别称取活性小分子和soluplus,经过研钵充分研磨混合。
[0039]
用10ml纯净水分别溶解过量的无定型药物组合物前体粉末和物理混合物粉末,在37℃下120r/min的摇床中震荡30分钟,为保证每个样品溶液都处于过饱和状态,震荡过程中始终保证溶液中有未溶解的固体。震荡30分钟后取上清液,过0.22μm的微孔滤膜,适当稀释后经hplc测定无定型药物组合物前体粉末溶解在水中的浓度,见表1。结果显示无定型药物组合物前体显著提升此类化合物在水为媒介的溶液中溶解度,实现此类化合物有效药用浓度1-5mg/ml的药物制备和应用需求。
[0040]
表1无定型药物组合物前体提升式1-式5化合物溶解度结果
[0041][0042][0043]
实施例2
[0044]
本实施例的无定型药物组合物前体粉末主要原料由式4化合物4和soluplus(1:25)组成,其具体的制备工艺如下:将式4化合物4和soluplus均匀混合溶解于20%四氢呋喃-丙酮溶液(v/v)中,室温充分搅拌30分钟,将混合液通过旋转蒸发仪在50℃下去除溶剂,得到式4化合物4无定型药物组合物前体粉末。
[0045]
实施例3
[0046]
本实施例的无定型药物组合物前体粉末主要原料由式4化合物4和soluplus(1:15)组成,其具体的制备工艺如下:将式4化合物4和soluplus均匀混合溶解于20%丙酮-乙腈溶液(v/v)中,在35℃下充分搅拌30分钟,将混合物溶液用喷雾干燥法进行干燥,喷雾干燥参数设置为:入口温度70℃、出口温度60℃、雾化气流量400l/h、蠕动泵速度为2ml/min,收集粉末,即得式4化合物4无定型药物组合物前体粉末。
[0047]
实施例4
[0048]
本实施例的无定型药物组合物前体粉末主要原料由式4化合物4和soluplus(1:30)组成,其具体的制备工艺如下:将式4化合物4和soluplus均匀混合,溶解于丙酮中,在35℃下充分搅拌30分钟,将混合物溶液用喷雾干燥法进行干燥,喷雾干燥参数设置为:入口温度80℃、出口温度65℃、雾化气流量410l/h、蠕动泵速度为4ml/min,收集粉末,即得式4化合物4无定型药物组合物前体粉末。
[0049]
实施例5
[0050]
本实施例的无定型药物组合物前体粉末主要原料由式4化合物4和soluplus(1:
15)组成,其具体的制备工艺如下:将式4化合物4和soluplus均匀混合溶于二氯甲烷中,室温充分搅拌30分钟溶液澄清,将混合液通过旋转蒸发仪在30℃下去除溶剂,得到式4化合物4无定型药物组合物前体粉末。
[0051]
实施例6
[0052]
本实施例的无定型药物组合物前体粉末主要原料由式4化合物4和soluplus(1:10)组成,其具体的制备工艺如下:将式4化合物4和soluplus均匀混合溶于二氯甲烷中,室温充分搅拌30分钟溶液澄清,将混合液通过旋转蒸发仪在30℃下去除溶剂,得到式4化合物4无定型药物组合物前体粉末。
[0053]
实施例7
[0054]
实施例1-6所制得的式4化合物4的无定型药物组合物前体粉末的溶出度、表征、生物利用度实验研究,方法和结果如下。
[0055]
(一)溶出和释放度实验
[0056]
取实施例1-6的式4化合物4的无定型药物组合物前体粉末适量(约相当于化合物540mg)和等量纯化合物,直接投入溶出杯中,照溶出度与释放度测定法(chp2015版四部通则0931)第一法(篮法),以水900ml为溶出介质,转速为100r/min,溶出液温度为(37
±
0.5℃),经15,30,45,60,90,120,150,180min分别取溶液5ml,同时补充等体积的溶出介质,用0.22μm微孔滤膜滤过,通过hplc测定药物浓度。
[0057]
其结果如图1所示,本发明所制备的实施例1-6的无定型药物组合物前体粉末均显著提升了化合物4在水中的溶出和释放率,实施例5制备的无定型药物组合物前体粉末溶解释放活性分子表现最优,然而实施例1-6不同组方和工艺条件制备的化合物4的无定型药物组合物前体粉末在水中的溶出和释放率略有不同,活性分子和soluplus的比例以及制备过程的工艺参数都能影响无定型药物组合物前体粉末中药物溶解释放的速度和程度,本发明采用添加羟丙基甲基纤维素邻苯二甲酸酯的手段解决活性分子释放的不稳定问题,同时提升无定型药物组合物前体粉末的贮藏稳定性,见实施例8,9,10,11。
[0058]
(二)实施例5的无定型药物组合物前体粉末表征
[0059]
(1)x射线衍射(xrd)分析:
[0060]
对本发明实施例5制备得到的化合物4无定型药物组合物前体粉末以及其原料、载体、物理混合物进行xrd分析。图2中(a)是单独化合物4的xrd扫描图,可以看到有多个特征衍射峰;(b)是载体soluplus的xrd扫描图,无特征衍射峰;(c)是化合物4和载体soluplus物理混合物的xrd扫描图,可以看到仍存在多个化合物4的特征衍射峰,说明化合物4仍以晶型的形式存在;(d)是实施例5制备得到的化合物4无定型药物组合物前体粉末的xrd扫描图,无特征衍射峰,说明化合物4以无定型状态存在于无定型药物组合物前体粉末中。
[0061]
(2)差示扫描量热法(dsc)分析:
[0062]
对本发明实施例5制备得到的化合物4无定型药物组合物前体粉末以及其原料、载体、物理混合物进行dsc分析。图3中(a)是化合物4的dsc扫描图,在280℃左右存在明显的吸热峰;(b)是载体soluplus的dsc扫描图,在70℃和320℃左右有明显的吸热峰;(c)是化合物4和载体soluplus物理混合物的dsc扫描图,与(b)相比在280℃左右出现轻微的化合物4的吸热峰,这表明化合物4与载体soluplus物理混合后仍以晶型形式存在;(d)是实施例5制备得到的化合物4无定型药物组合物前体粉末的dsc扫描图,可以明显看出化合物4的吸热峰
消失,载体soluplus在70℃的吸热峰向右偏移,320℃的吸热峰向左偏移且增强,表明化合物4和载体soluplus发生作用,以无定型状态存在于无定型药物组合物前体粉末中。
[0063]
(三)靶细胞摄取吸收实验
[0064]
靶细胞摄取吸收小分子药物实验采用结肠癌ht-29、sw1116/hcpt耐药细胞,胰腺癌panc-1、mia paca-2四种细胞株,开展化合物4的3组不同配制方式的细胞摄取实验,结果见图4。图4显示在四种靶细胞上实施例5制备得到的化合物4无定型药物组合物前体粉末能够实现靶细胞对化合物4摄取率较常规配制和环糊精增溶法显著提升。
[0065]
化合物4的3组不同配制方式:
[0066]
化合物4常规配制工作液:化合物4的dmso储备液(500μm)用含1%fbs的培养基稀释成1μm的工作液;
[0067]
实施例5的无定型药物组合物前体粉末工作液:0.6mg/ml化合物4的无定型药物组合物前体粉末用含1%fbs的培养基稀释成1μm的工作液。
[0068]
化合物4+环糊精工作液:按照dr li fengzhi专利公开方法配制化合物4和环糊精、dmso、peg混合液备用。
[0069]
(四)化合物4无定型药物组合物前体粉末生物利用实验
[0070]
针对4种活性分子处理形式开展药代动力学实验,
①
按照dr li fengzhi专利公开方法配制化合物4和环糊精、dmso、peg混悬液;
②
本发明实施例5制备得到的化合物4无定型药物组合物前体粉末(1:15);
③
本发明实施例6制备得到的化合物4无定型药物组合物前体粉末(1:10);
④
化合物4和soluplus物理混合物(1:15),进行药代动力学药时曲线的测定,选择雌性sd大鼠,通过灌胃一次性给药后定时眼眶取血测定得到,hplc法分析血液中化合物4浓度,用das2.0软件对结果进行分析,主要的药动学参数以非房室模型计算,得到的药时曲线结果如图5所示,各项药代动力学参数如表2所示。
[0071]
实施例5和实施例6制备的无定型药物组合物前体粉末在大鼠体内生物利用度显著优于dr li fengzhi等学者报道的环糊精混悬液技术组,实施例5的生物利用度最高,auc
0-72h
值明显高于其他3组,实施例5(1:15)的生物利用度是实施例6(1:10)的3.09倍,这与实施例6的无定型药物组合物前体粉末溶出释放活性分子慢有关(图1)。物理混合(1:15)组生物利用度较环糊精组也有提升,表明在sd大鼠体内环境下soluplus形成的胶束溶液促进化合物4溶解和吸收。
[0072]
表2灌胃给药实施例5和实施例6(8mg/kg化合物4)后的药时曲线
[0073][0074]
注:与环糊精混悬液组比较,ap《0.0001
[0075]
从实施例1-7可得出,本发明所公开的fl118及其7位结构修饰衍生物的无定型药
物组合物前体粉末制备方法,可以显著提升此类化合物溶解度,充分满足此类化合物有效药用浓度1-5mg/ml的药用制药需求。但是无定型药物组合物前体粉末的药物溶出释放程度是影响活性分子吸收利用发挥药效的关键,实施例7中显示不同组方和工艺条件制备的化合物4的无定型药物组合物前体粉末(实施例1-6)在水中的溶出和释放情况略有不同,影响到活性分子的吸收利用度不同。本发明采用添加羟丙基甲基纤维素邻苯二甲酸酯的手段增强活性分子释放率,同时增加无定型药物组合物前体粉末贮藏过程的稳定性。
[0076]
实施例8
[0077]
称取实施例1所制备的化合物4:soluplus(1:20)无定型前体100mg和羟丙基甲基纤维素邻苯二甲酸酯5mg(20:1),置于离心管内,涡旋5min至二者混合均匀。从而获化合物4,soluplus和羟丙基甲基纤维素邻苯二甲酸酯的药物组合物。实施例8制备的药物组合物室温放置6个月,检测其溶出释放率,结果如图6所示,从图可判断羟丙基甲基纤维素邻苯二甲酸酯的加入所得的实施例8药物组合物较实施例1的无定型前体溶出释放率高,而且室温放置6个月后实施例8药物组合物活性分子仍保持较高溶出释放率。
[0078]
实施例9
[0079]
称取实施例2所制备的化合物4:soluplus(1:25)无定型前体100mg和羟丙基甲基纤维素邻苯二甲酸酯5mg(20:1),置于离心管内,涡旋5min至二者混合均匀。从而获化合物4,soluplus和羟丙基甲基纤维素邻苯二甲酸酯的药物组合物。实施例9制备的药物组合物室温放置6个月,检测其溶出释放率,结果如图7所示,从图可判断羟丙基甲基纤维素邻苯二甲酸酯的加入所得的实施例9药物组合物较实施例2的无定型前体溶出释放率高,而且室温放置6个月后实施例9药物组合物活性分子仍保持较高溶出释放率。
[0080]
实施例10
[0081]
称取实施例5所制备的化合物4:soluplus(1:15)无定型前体100mg和羟丙基甲基纤维素邻苯二甲酸酯8mg(25:2),置于离心管内,涡旋5min至二者混合均匀。从而获化合物4,soluplus和羟丙基甲基纤维素邻苯二甲酸酯的药物组合物。实施例5制备的药物组合物室温放置6个月,检测其溶出释放率,结果如图8所示,从图可判断羟丙基甲基纤维素邻苯二甲酸酯的加入所得的实施例10药物组合物较实施例5的无定型前体溶出释放率高,而且室温放置6个月后实施例10药物组合物活性分子仍保持较高溶出释放率。
[0082]
实施例11
[0083]
称取实施例6所制备的化合物4:soluplus(1:10)无定型前体100mg和羟丙基甲基纤维素邻苯二甲酸酯10mg(10:1),置于离心管内,涡旋5min至二者混合均匀。从而获化合物4,soluplus和羟丙基甲基纤维素邻苯二甲酸酯的药物组合物。实施例6制备的药物组合物室温放置6个月,检测其溶出释放率,结果如图9所示,从图可判断羟丙基甲基纤维素邻苯二甲酸酯的加入所得的实施例11药物组合物较实施例6的无定型前体溶出释放率高,而且室温放置6个月后实施例11药物组合物活性分子仍保持相对较高溶出释放率。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1