一种基于多头注意力的蛋白质翻译后修饰预测方法
1.本发明属于生物信息技术领域,尤其涉及一种基于多头注意力的蛋白质翻译后修饰预测方法。
背景技术:
2.通过生物理化实验预测蛋白质序列上氨基酸是否发生了翻译后修饰,需要在对实验样本预处理之后进行蛋白质提取以及分级分离,对待检测肽段进行富集,然后对富集之后的肽段进行质谱分析。这种方式时间成本和经济成本都较大,不适用于繁多的修饰翻译后预测。
3.尽管现有的一些预测蛋白质序列上氨基酸翻译后修饰方法通过计算的方法实现蛋白质序列的预测,在效率和成本开销方面相对于通过生物学实验方法来确定酰化位点是一个格外出众的优点,但这些计算方法往往需要较为丰富的生物学背景知识,来从原始的蛋白质序列中按照生物学特征提取方法构造特征。
4.采用现有的蛋白质序列中氨基酸翻译后修饰预测方法存在以下问题:对蛋白质序列的处理严重依赖于生物学背景的特征工程,也忽视了蛋白质序列中的多肽组成信息。
技术实现要素:
5.针对现有技术中的上述不足,本发明提供的一种基于多头注意力的蛋白质翻译后修饰预测方法,通过多尺度词向量全面利用原始蛋白质序列中的多肽组成信息,并降低了对于生物学特征工程的依赖。
6.为了达到上述发明目的,本发明采用的技术方案为:
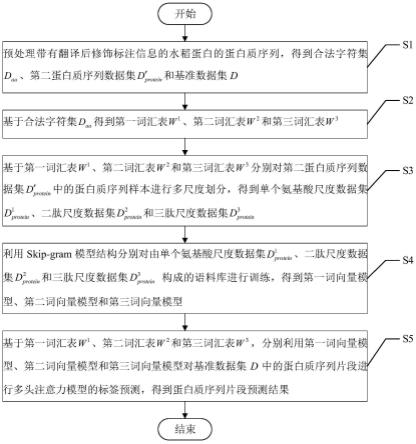
7.本发明提供一种基于多头注意力的蛋白质翻译后修饰预测方法,包括如下步骤:
8.s1、预处理带有翻译后修饰标注信息的蛋白质序列,得到合法字符集d
aa
、第二蛋白质序列数据集d
′
protein
和基准数据集d;
9.s2、基于合法字符集d
aa
得到第一词汇表w1、第二词汇表w2和第三词汇表w3;
10.s3、基于第一词汇表w1、第二词汇表w2和第三词汇表w3分别对第二蛋白质序列数据集d
′
protein
中的蛋白质序列样本进行多尺度划分,得到单个氨基酸尺度数据集二肽尺度数据集和三肽尺度数据集
11.s4、利用skip-gram模型结构分别对由单个氨基酸尺度数据集二肽尺度数据集和三肽尺度数据集构成的语料库进行训练,得到第一词向量模型、第二词向量模型和第三词向量模型;
12.s5、基于第一词汇表w1、第二词汇表w2和第三词汇表w3,分别利用第一词向量模型、第二词向量模型和第三词向量模型对基准数据集d中的蛋白质序列片段进行多头注意力模型的标签预测,得到蛋白质序列片段预测结果。
13.本发明的有益效果为:本发明提供的一种基于多头注意力的蛋白质翻译后修饰预测方法提取了蛋白序列中的单个氨基酸、二肽和三肽的多个尺度的信息,并通过三个通道提取各个尺度下的词向量中的特征,并结合通道注意力机制输出多个通道融合后的特征,提高了对蛋白上氨基酸翻译后修饰的预测效果;本方案从采集到的原始蛋白序列数据集中,按照三个尺度构造出,单氨基酸、二肽和三肽的三个尺度的数据集作为语料库,训练了多尺度词向量,只通过词向量作为原始序列的特征,不再依赖于传统生物学特征提取方法。
14.进一步地,所述步骤s1包括如下步骤:
15.s11、获取带有翻译后修饰标注信息的蛋白质序列集合;
16.s12、定义蛋白质序列集合中蛋白质序列样本的标注信息label的表达式:
17.label={pos1,pos2,
…
,posi,
…
,pos
l
}
18.其中,posi表示蛋白质序列样本的标注信息中第i个位点处发生了某种翻译后修饰,其中,i=1,2,
…
,l,l表示蛋白质序列样本中发生了某种翻译后修饰的位点总数;
19.s13、将蛋白质序列集合中包括不属于合法字符集d
aa
中氨基酸简称字符的蛋白质序列样本清除,得到第一蛋白质序列数据集d
protein
;
20.s14、通过cd-hit对蛋白质序列数据集d
protein
去冗余,得到第二蛋白质序列数据集d
′
protein
;
21.s15、对第二蛋白质序列数据集d
′
protein
中各蛋白质序列样本设置窗口半径为k
′
的滑动窗口,并利用滑动窗口将各蛋白质序列样本裁剪,得到若干长度均为2k
′
+1的蛋白质序列片段;
22.s16、根据蛋白质序列集合中各蛋白质序列样本的标注信息label,得到发生翻译后修饰的氨基酸位点集合,并判断各蛋白质序列片段中心处氨基酸是否处于发生翻译后修饰的氨基酸位点集合,若是则将对应蛋白质序列片段置于正样本数据集中,否则将对应蛋白质序列片段置于负样本数据集中;
23.s17、通过随机采样从负样本数据集中获取与正样本数据集中蛋白质序列片段数量一致的蛋白质序列片段,并将获取得到的蛋白质序列片段与正样本数据集合并,得到基准数据集d。
24.采用上述进一步方案的有益效果为:提供对原始蛋白序列预处理的方法,得到合法字符集d
aa
、第二蛋白质序列数据集d
′
protein
和基准数据集d,为得到词汇表和尺度数据集提供基础。
25.进一步地,所述蛋白质序列集合中的蛋白质序列样本由组成该序列的氨基酸的字母简称表示,并以生物体内二十种常见的氨基酸简称字符g、a、v、l、i、p、f、y、w、s、t、c、m、r和h的集合作为合法字符集d
aa
。
26.采用上述进一步方案的有益效果为:提供合法字符集的范围以及蛋白质序列样本的表示方法。
27.进一步地,所述步骤s15包括如下步骤:
28.s151、对第二蛋白质序列数据集d
′
protein
中各蛋白质序列样本设置窗口半径为k
′
的滑动窗口;
29.s152、将滑动窗口在对应的蛋白质序列样本上滑动;
30.s153、选取窗口内中心点处氨基酸两侧长度为k
′
的序列进行裁剪,得到若干长度
均为2k
′
+1的蛋白质序列片段。
31.采用上述进一步方案的有益效果为:提供滑动窗口裁剪等长蛋白质序列片段的方法,为得到基准数据集d提供基础。
32.进一步地,所述步骤s2包括如下步骤:
33.s21、分别构建第一词汇表w1、第二词汇表w2和第三词汇表w3,并将第一词汇表w1、第二词汇表w2和第三词汇表w3均初始化为空集;
34.s22、基于合法字符集d
aa
中的二十种氨基酸,分别以单个氨基酸、二肽和三肽三个尺度进行构词;
35.s23、将单个氨基酸构成的词置于第一词汇表w1中;
36.s24、将两个氨基酸组成的二肽构成的词置于第二词汇表w2中;
37.s25、将三个氨基酸组成的三肽构成的词置于第三词汇表w3中。
38.采用上述进一步方案的有益效果为:提供第一词汇表w1、第二词汇表w2和第三词汇表w3具体的构建方法,为得到尺度数据集和实现蛋白质序列片段预测提供基础。
39.进一步地,所述步骤s3包括如下步骤:
40.s31、将第二蛋白质序列数据集d
′
protein
中的各蛋白质序列样本通过氨基酸组成进行表示,得到氨基酸组成表示的蛋白质序列样本seq:
41.seq=[aa1,aa2,
…
,aaii′
,
…
,aan]
[0042]
seq∈d
′
protein
,aai′
∈d
aa
[0043]
其中,aai′
表示氨基酸组成表示的蛋白质序列样本seq中第i
′
个位置处的氨基酸,其中,i
′
=1,2,3,
…
,n,n表示氨基酸组成表示的蛋白质序列样本seq的氨基酸总数;
[0044]
s32、分别以单个氨基酸、二肤和三肤三个尺度对氨基酸组成表示的蛋白质序列样本seq进行划分,得到分别由第一词汇表w1、第二词汇表w2和第三词汇表w3中的词构成的单氨基酸序列seq1、二肤序列seq2和三肤序列seq3:
[0045]
seq1=[aa1,aa2,
…
,aai′
,
…
,aan]=seq
[0046]
seq2=[(aa1aa2),(aa2aa3),
…
,(aai′
aai′
+1
),
…
,(aa
n-1
aan)]
[0047]
seq3=[(aa1aa2aa3),(aa2aa3aa4),
…
,(aai′
aai′
+1
aai′
+2
),
…
,(aa
n-2
aa
n-1
aan)]
[0048]
aai′
∈w1,aai′
aai′
+1
∈w2,aai′
aai′
+1
aai′
+2
∈w3[0049]
其中,aa
′i′
表示通过第一词汇表w1中的词表示的单氨基酸序列seq1中第i
′
个位置处的氨基酸,aai′
aai′
+1
表示通过第二词汇表w2中的词表示的二肽序列seq2中第i
′
个位置处的二肽,aai′
aai′
+1
aai′
+2
表示通过第三词汇表w3中的词表示的三肽序列seq3中第i
′
个位置处的三肽,其中,i
′
=1,2,3,
…
,n;
[0050]
s33、依次对第二蛋白质序列数据集d
′
protein
中的蛋白质序列样本seq分别进行单个氨基酸、二肽和三肽三个尺度的划分,得到单个氨基酸尺度数据集二肽尺度数据集和三肽尺度数据集
[0051]
采用上述进一步方案的有益效果为:提供对第二蛋白质序列数据集d
′
protein
中的蛋白质序列样本进行多尺度划分,得到单个氨基酸尺度数据集二肽尺度数据集和三肽尺度数据集的方法,为得到词向量模型和进行蛋白质序列片段预测提
供基础。
[0052]
进一步地,所述步骤s5包括如下步骤:
[0053]
s51、将基准数据集d中各蛋白质序列片段分别进行单个氨基酸、二肽和三肽三个尺度的划分;
[0054]
s52、分别利用第一词汇表w1、第二词汇表w2和第三词汇表w3中的词对三个尺度划分后的蛋白质序列片段重构,得到若干氨基酸样本片段序列若干二肽样本片段序列和若干三肽样本片段序列其中,i
″
表示基准数据集中第i
″
个蛋白质序列片段;
[0055]
s53、利用第一词向量模型将各氨基酸样本片段序列转换为氨基酸样本片段词向量,利用第二词向量模型将各二肽样本片段序列转换为二肽样本片段词向量,利用第三词向量模型将各三肽样本片段序列转换为三肽样本片段词向量;
[0056]
s54、基于氨基酸样本片段词向量构建氨基酸样本片段词向量矩阵基于二肽样本片段词向量构建二肽样本片段词向量矩阵基于三肽样本片段词向量构建三肽样本片段词向量矩阵
[0057]
s55、利用旋转位置编码模块rope分别对各氨基酸样本片段词向量、各二肽样本片段词向量和各三肽样本片段词向量添加位置编码信息,得到添加位置编码后的氨基酸样本片段词向量矩阵二肽样本片段词向量矩阵和三肽样本片段词向量矩阵
[0058]
s56、将添加位置编码后的氨基酸样本片段词向量矩阵输入单个氨基酸序列通道的多头注意力模块multi-att中,将二肽样本片段词向量矩阵输入二肽序列通道的多头注意力模块multi-att中,将三肽样本片段词向量矩阵输入三肽序列通道的多头注意力模块multi-att中,并通过各通道中的多头注意力模块学习得到三种尺度下的词向量矩阵特征:
[0059][0060]
其中,表示j通道中对第i
″
个蛋白质序列片段提取到的词向量矩阵特征,其中,j=1时表示单个氨基酸序列通道,j=2时表示二肽序列通道,j=3时表示三肽序列通道;
[0061]
s57、将单个氨基酸序列通道对第i
″
个蛋白质序列片段提取到的词向量矩阵特征、二肽序列通道对第i
″
个蛋白质序列片段提取到的词向量矩阵特征和三肽序列通道对第i
″
个蛋白质序列片段提取到的词向量矩阵特征进行拼接,并将拼接结果输入注意力模块chanel-att中学习三肽序列通道的注意力权重,得到通道注意力权重调整后的第i
″
个蛋白质序列片段特征:
[0062][0063]
其中,oi″
表示通道注意力权重调整后的第i
″
个蛋白质序列片段特征;
[0064]
s58、将通道注意力权重调整后的第i
″
个蛋白质序列片段特征输入全连接层,得到蛋白质序列片段预测结果:
[0065]
labeli″
=soft max(w
·
oi″
+b)
[0066]
其中,labeli″
表示第i
″
个蛋白质序列片段的预测标签,w和b分别表示最后一层线性层网络的神经元权重矩阵和偏置,其中,当第i
″
个蛋白质序列片段为正样本数据时其预测标签为1,当第i
″
个蛋白质序列片段为负样本数据时其预测标签为0。
[0067]
采用上述进一步方案的有益效果为:提供对各蛋白质序列片段进行氨基酸翻译后修饰预测的方法,并通过多尺度词向量全面利用原始蛋白质序列中的多肽组成信息,并降低了对于生物学特征工程的依赖。
附图说明
[0068]
图1为本发明实施例中一种基于多头注意力的蛋白质翻译后修饰预测方法的步骤图。
具体实施方式
[0069]
下面对本发明的具体实施方式进行描述,以便于本技术领域的技术人员理解本发明,但应该清楚,本发明不限于具体实施方式的范围,对本技术领域的普通技术人员来讲,只要各种变化在所附的权利要求限定和确定的本发明的精神和范围内,这些变化是显而易见的,一切利用本发明构思的发明创造均在保护之列。
[0070]
如图1所示,在本发明的一个实施例中,本发明提供一种基于多头注意力的蛋白质翻译后修饰预测方法,包括如下步骤:
[0071]
目前提供有蛋白质上氨基酸翻译后修饰问题的数据开源平台有ptm viewer,但其中只有数量不多的样本不能满足需求;因此,本实施例还从已发表的实验论文中采集到存在翻译后修饰现象的蛋白质uniport id,以及该蛋白质上发生ptm的氨基酸的坐标信息;然后通过蛋白质uniport id在uniport数据库中检索到该蛋白质完整的氨基酸序列;结合氨基酸序列以及其上发生的ptm位置,便构成了一条存在着一个或多个ptm位点的原始样本集合a;将从开源数据平台ptm viewer中获得的数据作为原始样本集合b;为了确保数据集中样本的正确性,方案按照合法字符集d
aa
将原始样本集合a和原始样本集合b中的错误样本进行了筛选;筛选之后将原始样本集合a和原始样本集合b中的合法样本合并在一起,构成原始数据集d
raw
;
[0072]
s1、预处理带有翻译后修饰标注信息的蛋白质序列,得到合法字符集d
aa
、第二蛋白质序列数据集d
′
protein
和基准数据集d;
[0073]
所述步骤s1包括如下步骤:
[0074]
s11、基于原始数据集d
raw
获取带有翻译后修饰标注信息的蛋白质序列集合;
[0075]
s12、定义蛋白质序列集合中蛋白质序列样本的标注信息label的表达式:
[0076]
label={pos1,pos2,
…
,posi,
…
,pos
l
}
[0077]
其中,posi表示蛋白质序列样本的标注信息中第i个位点处发生了某种翻译后修饰,其中,i=1,2,
…
,l,l表示蛋白质序列样本中发生了某种翻译后修饰的位点总数;
[0078]
s13、将蛋白质序列集合中包括不属于合法字符集d
aa
中氨基酸简称字符的蛋白质序列样本清除,得到第一蛋白质序列数据集d
protein
;
[0079]
所述蛋白质序列集合中的蛋白质序列样本由组成该序列的氨基酸的字母简称表
示,并以生物体内二十种常见的氨基酸简称字符g(甘氨酸)、a(丙氨酸)、v(缬氨酸)、l(亮氨酸)、i(异亮氨酸)、p(脯氨酸)、f(苯丙氨酸)、y(酪氨酸)、w(色氨酸)、s(丝氨酸)、t(苏氨酸)、c(半胱氨酸)、m(蛋氨酸)、n(天冬酰胺)、q(谷氨酰胺)、d(天冬氨酸)、e(谷氨酸)、k(赖氨酸)、r(精氨酸)和h(组氨酸)的集合作为合法字符集d
aa
;
[0080]
s14、通过cd-hit对蛋白质序列数据集d
protein
去冗余,得到第二蛋白质序列数据集d
′
protein
,本实施例中使用cd-hit选择的去冗余参数为0.3;
[0081]
s15、对第二蛋白质序列数据集d
′
protein
中各蛋白质序列样本设置窗口半径为k
′
的滑动窗口,并利用滑动窗口将各蛋白质序列样本裁剪,得到若干长度均为2k
′
+1的蛋白质序列片段,本实施例中设滑动窗口半径为k
′
=25,则蛋白质序列片段长度为51;
[0082]
所述步骤s15包括如下步骤:
[0083]
s151、对第二蛋白质序列数据集d
′
protein
中各蛋白质序列样本设置窗口半径为k
′
的滑动窗口;
[0084]
s152、将滑动窗口在对应的蛋白质序列样本上滑动;
[0085]
s153、选取窗口内中心点处氨基酸两侧长度为k
′
的序列进行裁剪,得到若干长度均为2k
′
+1的蛋白质序列片段;
[0086]
s16、根据蛋白质序列集合中各蛋白质序列样本的标注信息label,得到发生翻译后修饰的氨基酸位点集合,并判断各蛋白质序列片段中心处氨基酸是否处于发生翻译后修饰的氨基酸位点集合,若是则将对应蛋白质序列片段置于正样本数据集中,否则将对应蛋白质序列片段置于负样本数据集中;
[0087]
s17、通过随机采样从负样本数据集中获取与正样本数据集中蛋白质序列片段数量一致的蛋白质序列片段,并将获取得到的蛋白质序列片段与正样本数据集合并,得到基准数据集d;本实施例通过随机抽样,使数据集中的正负样本数据达到均衡;
[0088]
s2、基于合法字符集d
aa
得到第一词汇表w1、第二词汇表w2和第三词汇表w3;
[0089]
所述步骤s2包括如下步骤:
[0090]
s21、分别构建第一词汇表w1、第二词汇表w2和第三词汇表w3,并将第一词汇表w1、第二词汇表w2和第三词汇表w3均初始化为空集;
[0091]
s22、基于合法字符集d
aa
中的二十种氨基酸,分别以单个氨基酸、二肽和三肽三个尺度进行构词;
[0092]
s23、将单个氨基酸构成的词置于第一词汇表w1中,其词汇表大小为20;
[0093]
s24、将两个氨基酸组成的二肽构成的词置于第二词汇表w2中,其词汇表大小为400;
[0094]
s25、将三个氨基酸组成的三肽构成的词置于第三词汇表w3中,其词汇表大小为8000;
[0095]
s3、基于第一词汇表w1、第二词汇表w2和第三词汇表w3分别对第二蛋白质序列数据集d
′
protein
中的蛋白质序列样本进行多尺度划分,得到单个氨基酸尺度数据集二肽尺度数据集和三肽尺度数据集
[0096]
所述步骤s3包括如下步骤:
[0097]
s31、将第二蛋白质序列数据集d
′
protein
中的各蛋白质序列样本通过氨基酸组成进
行表示,得到氨基酸组成表示的蛋白质序列样本seq:
[0098]
seq=[aa1,aa2,
…
,aai′
,
…
,aan]
[0099]
seq∈d
′
protein
,aai′
∈d
aa
[0100]
其中,aai′
表示氨基酸组成表示的蛋白质序列样本seq中第i
′
个位置处的氨基酸,其中,i
′
=1,2,3,
…
,n,n表示氨基酸组成表示的蛋白质序列样本seq的氨基酸总数;
[0101]
s32、分别以单个氨基酸、二肽和三肽三个尺度对氨基酸组成表示的蛋白质序列样本seq进行划分,得到分别由第一词汇表w1、第二词汇表w2和第三词汇表w3中的词构成的单氨基酸序列seq1、二肽序列seq2和三肽序列seq3:
[0102]
seq1=[aa1,aa2,
…
,aai′
,
…
,aan]=seq
[0103]
seq2=[(aa1aa2),(aa2aa3),
…
,(aai′
aai′
+1
),
…
,(aa
n-1
aan)]
[0104]
seq3=[(aa1aa2aa3),(aa2aa3aa4),
…
,(aai′
aai′
+1
aai′
+2
),
…
,(aa
n-2
aa
n-1
aan)]
[0105]
aai′
∈w1,aai′
aai′
+1
∈w2,aai′
aai′
+1
aai′
+2
∈w3[0106]
其中,aa
′i′
表示通过第一词汇表w1中的词表示的单氨基酸序列seq1中第i
′
个位置处的氨基酸,aai′
aai′
+1
表示通过第二词汇表w2中的词表示的二肽序列seq2中第i
′
个位置处的二肽,aai′
aai′
+1
aai′
+2
表示通过第三词汇表w3中的词表示的三肽序列seq3中第i
′
个位置处的三肽,其中,i
′
=1,2,3,
…
,n;
[0107]
对于经过上述多尺度划分之后均可得到三份序列seq1、seq2和seq3,长度分别为n、n-1和n-2,经过填充,将这些序列长度均调整为n,这些序列可以组成三份由不同尺度词汇表构成的新数据集,分别为单个氨基酸尺度数据集二肽尺度数据集和三肽尺度数据集且三个新数据集的规模均等同于原数据集规模|d
protein
|;
[0108]
s33、依次对第二蛋白质序列数据集d
′
protein
中的蛋白质序列样本seq分别进行单个氨基酸、二肽和三肽三个尺度的划分,得到单个氨基酸尺度数据集二肽尺度数据集和三肽尺度数据集
[0109]
s4、利用skip-gram模型结构分别对由单个氨基酸尺度数据集二肽尺度数据集和三肽尺度数据集构成的语料库进行训练,得到第一词向量模型、第二词向量模型和第三词向量模型;本实施例中训练出的三个尺度的氨基酸词向量维度均别为100;将训练得到的词向量用于第一词汇表w1、第二词汇表w2和第三词汇表w3中对应的多尺度氨基酸词,只有在语料库中出现的多尺度氨基酸词才能够学习到词向量,其他未在语料库中出现的词汇默认初始化为100维的0向量,不过在模型的氨基酸词embedding中并不会用到那些默认初始化的氨基酸词向量,因此本实施例中这样初始化并不会影响到对词向量的使用;
[0110]
s5、基于第一词汇表w1、第二词汇表w2和第三词汇表w3,分别利用第一词向量模型、第二词向量模型和第三词向量模型对基准数据集d中的蛋白质序列片段进行多头注意力模型的标签预测,得到蛋白质序列片段预测结果;
[0111]
所述步骤s5包括如下步骤:
[0112]
s51、将基准数据集d中各蛋白质序列片段分别进行单个氨基酸、二肽和三肽三个
尺度的划分;
[0113]
s52、分别利用第一词汇表w1、第二词汇表w2和第三词汇表w3中的词对三个尺度划分后的蛋白质序列片段重构,得到若干氨基酸样本片段序列若干二肽样本片段序列和若干三肽样本片段序列其中,i
″
表示基准数据集中第i
″
个蛋白质序列片段;
[0114]
本实施例中氨基酸样本片段序列二肽样本片段序列和三肽样本片段序列的维度分别为1
×
51、1
×
50和1
×
49并将三者padding为1
×
51的序列,然后按照词汇表第一词汇表w1、第二词汇表w2和第三词汇表w3中词向量表达;
[0115]
s53、利用第一词向量模型将各氨基酸样本片段序列转换为氨基酸样本片段词向量,利用第二词向量模型将各二肽样本片段序列转换为二肽样本片段词向量,利用第三词向量模型将各三肽样本片段序列转换为三肽样本片段词向量;
[0116]
s54、基于氨基酸样本片段词向量构建氨基酸样本片段词向量矩阵基于二肽样本片段词向量构建二肽样本片段词向量矩阵基于三肽样本片段词向量构建三肽样本片段词向量矩阵
[0117]
s55、利用旋转位置编码模块rope分别对各氨基酸样本片段词向量、各二肽样本片段词向量和各三肽样本片段词向量添加位置编码信息,得到添加位置编码后的氨基酸样本片段词向量矩阵二肽样本片段词向量矩阵和三肽样本片段词向量矩阵所述氨基酸样本片段词向量矩阵二肽样本片段词向量矩阵和三肽样本片段词向量矩阵的维度均为1
×
51
×
100;
[0118]
s56、将添加位置编码后的氨基酸样本片段词向量矩阵输入单个氨基酸序列通道的多头注意力模块multi-att中,将二肽样本片段词向量矩阵输入二肽序列通道的多头注意力模块multi-att中,将三肽样本片段词向量矩阵输入三肽序列通道的多头注意力模块multi-att中,并通过各通道中的多头注意力模块学习得到三种尺度下的词向量矩阵特征:
[0119][0120]
其中,表示j通道中对第i
″
个蛋白质序列片段提取到的特征,其维度为1
×
51
×
100,其中,j=1时表示单个氨基酸序列通道,j=2时表示二肽序列通道,j=3时表示三肽序列通道;
[0121]
s57、将单个氨基酸序列通道对第i
″
个蛋白质序列片段提取到的特征、二肽序列通道对第i
″
个蛋白质序列片段提取到的特征和三肽序列通道对第i
″
个蛋白质序列片段提取到的特征拼接后输入注意力模块chanel-att中学习三肽序列通道的注意力权重,得到通道注意力权重调整后的第i
″
个蛋白质序列片段特征:
[0122][0123]
其中,oi″
表示通道注意力权重调整后的第i
″
个蛋白质序列片段特征,其维度均为1
×
51
×
100;
[0124]
s58、将通道注意力权重调整后的第i
″
个蛋白质序列片段特征输入全连接层,得到蛋白质序列片段预测结果:
[0125]
labeli″
=soft max(w
·
oi″
+b)
[0126]
其中,labeli″
表示第i
″
个蛋白质序列片段的预测标签,w和b分别表示最后一层线性层网络的神经元权重矩阵和偏置,其中,当第i
″
个蛋白质序列片段为正样本数据时其预测标签为1,当第i
″
个蛋白质序列片段为负样本数据时其预测标签为0。
[0127]
为了验证本发明的蛋白质上氨基酸翻译后修饰问题的预测准确率的优越性,我们对本发明方法与基于卷积神经网络(cnn)的方法进行了对比试验;实验数据集共有754个正样本和随机抽样的等量正样本,共有1508条,试验情况如表1所示:
[0128]
表1
[0129][0130]
由表1可得,本专利方法的预测性能比现有方法提升了13%左右,取得了较大的提高。
[0131]
本发明分成单氨基酸、二肽和三肽三个尺度从蛋白质序列中获得多尺度词向量,然后经过旋转位置编码添加位置信息之后,分为三个通道基于多头注意力从中三个尺度的词向量中提取特征;再经过通道注意力对三个通道特征的权重进行调整,最后将三个通道的融合后的特征通过一层线性层实现对输入序列的标签预测,从而实现了结合蛋白质序列中氨基酸、二肽和三肽的多个尺度序列组成信息的结合,提高了对蛋白上氨基酸翻译后修饰的预测性能。
[0132]
本发明的有益效果为:
[0133]
(1)、本发明提取了蛋白序列中的单个氨基酸、二肽和三肽的多个尺度的信息,通过三个通道提取各个尺度下的词向量中的特征,并结合通道注意力机制输出多个通道融合后的特征,提高了对蛋白上氨基酸翻译后修饰的预测效果。
[0134]
(2)、本发明从采集到的原始蛋白序列数据集中,按照三个尺度构造出,单氨基酸、二肽和三肽的三个尺度的数据集作为语料库,训练了多尺度词向量,只通过词向量作为原始序列的特征,不再依赖于传统生物学特征提取方法,解决了现有方法实现对水蛋白质上氨基酸翻译后修饰问题预测时对生物学特征提取方法的依赖性。
[0135]
尽管上面对本发明说明性的具体实施方式进行了描述,以便于本技术领域的技术人员理解本发明,但应该清楚,本发明不限于具体实施方式的范围,对本技术领域的普通技术人员来讲,只要各种变化在所附的权利要求限定和确定的本发明的精神和范围内,这些变化是显而易见的,一切利用本发明构思的发明创造均在保护之列。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1