一种前药、两性离子水凝胶及其制备方法、应用
1.本发明涉及高分子功能材料领域,具体涉及能够实现骨关节炎靶向治疗的透明质酸-对乙 酰氨基酚前药、基于该前药且能够在骨关节环境中缓慢释放该前药的两性离子水凝胶,以及 该两性离子水凝胶的制备方法和应用。
背景技术:
2.骨关节炎(oa)是最常见的关节炎形式,可以影响身体的较大和较小的关节,包括手、 脚、背部、髋部、膝盖和脊柱。骨关节炎会引起关节软骨的降解,使得关节软骨失去其力学 承载、关节润滑的功能,无论是健康关节还是病理环境下的关节,其中的关节软骨都是长期 处于剪切力的作用下。
3.为治疗骨关节炎,通常利用凝胶类产品放入关节环境中,持续向关节腔和周围组织提供 治疗剂以缓解、治疗骨关节炎。专利cn107427584a公开了一种关节脂肪垫制剂,其制剂可 以是凝胶、植入物、丝纤蛋白水凝胶、微球或纳米球,且优选采用包含透明质酸的凝胶,同 时,其治疗剂选自甾体抗炎剂、非甾体抗炎剂(nsaid)、抗炎性细胞因子、抗代谢物、n
‑ꢀ
甲基-d-天冬氨酸(nmda)受体拮抗剂、对乙酰氨基酚(扑热息痛)、阿片剂、环加氧酶-2(cox2) 抑制剂及其组合。该凝胶虽然能够在关节环境中持续释放治疗剂,然而,这类治疗剂在关节 环境中呈游离状,容易被带走,发挥的治疗效果有限。
4.不仅如此,传统的用于骨关节炎治疗的水凝胶类材料主要是通过剪切变稀作用实现其可 注射性,因此这种可注射材料如何在关节环境中应对普遍存在的剪切力同样成为亟待解决的 问题。
技术实现要素:
5.本发明的一个目的在于提供一种前药,该前药能够与两性离子单体反应、通过紫外光交 联接枝到两性离子水凝胶上,更加稳定地存在于关节腔内并缓慢地释放以提供更好的治疗效 果,同时通过透明质酸的靶向作用实现炎症的靶向治疗。
6.上述目的通过下述技术方案实现:
7.一种前药,所述前药具有式i所示的结构:
8.9.本技术方案中,将透明质酸钠和对乙酰氨基酚按照一定比例反应合成得到透明质酸-对乙 酰氨基酚(ha-pa)前药,其能够通过紫外光交联接枝到两性离子水凝胶上。透明质酸、对 乙酰氨基酚接枝在水凝胶上后,能够在关节环境中持续缓慢释放,相较于传统的小分子、游 离状的透明质酸、对乙酰氨基酚对骨关节炎的治疗效果更好。
10.在验证试验中,通过比较ha-pa前药、以及接枝有该前药的两性离子水凝胶的细胞毒性, 发现ha-pa前药与对应的水凝胶均具有良好的细胞相容性,两者对细胞的生长没有不利的影 响,并且,接枝在两性离子水凝胶上,不会影响两性离子水凝胶、透明质酸、对乙酰氨基酚 对关节的润滑和抗炎功能。
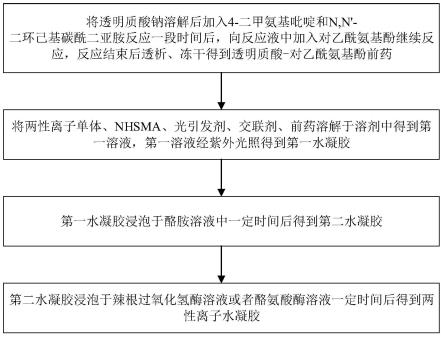
11.本发明的另一个目的在于提供一种基于前述任一种前药的两性离子水凝胶的制备方法, 该制备方法不会影响两性离子水凝胶和透明质酸的关节润滑和抗炎功能,还能够有效地提高 水凝胶的力学性能,同时,该制备方法易于实施,不需要特殊的设备,易于实现批量生产与 应用推广。
12.上述目的通过下述技术方案实现:
13.两性离子水凝胶的制备方法,包括以下步骤:
14.将两性离子单体、nhsma、光引发剂、交联剂、以及权利要求1所述的前药溶解于溶剂 中得到第一溶液,所述第一溶液经紫外光照得到第一水凝胶;
15.所述第一水凝胶浸泡于酪胺溶液中一定时间后得到第二水凝胶;
16.所述第二水凝胶浸泡于辣根过氧化氢酶溶液或者酪氨酸酶溶液一定时间后得到所述两性 离子水凝胶;
17.其中,所述两性离子单体为sbma、cbma或mpc。
18.本技术方案中,将ha-pa前药、两性离子单体、甲基丙烯酰n-羟基琥珀酰亚胺单体 (nhsma)、光引发剂和交联剂充分溶解于溶剂,例如去离子水或超纯水中,通过紫外光照 得到第一水凝胶,将ha-pa前药接枝在两性离子单体上。
19.接下来,将制备的第一水凝胶浸泡于酪胺溶液中一定时间后,浸泡于去离子水中一段时 间以去除未反应的酪胺分子。通过将第一水凝胶浸泡于酪胺溶液中,利用酪胺基团修饰第一 水凝胶得到酪胺修饰的第二水凝胶。随后,再将第二水凝胶浸泡于辣根过氧化氢酶(hrp) 溶液或者酪氨酸酶溶液中,得到表面具有辣根过氧化氢酶或酪氨酸酶的两性离子水凝胶。
20.当该两性离子水凝胶用于骨关节环境中时,由于骨关节环境中过氧化氢处于过表达状态, 浓度高于生理浓度,因此,骨关节环境中的过氧化氢能够与辣根过氧化氢酶或酪氨酸酶共同 作用,生物催化两性离子水凝胶上修饰的酪胺基团,从而使得两性离子水凝胶在关节腔内二 次酶交联以提高两性离子水凝胶的力学性能。
21.以两性离子单体为sbma为例,ha-pa前药接枝在sbma上后形成第一水凝胶(式ii), 第一水凝胶在浸泡了酪胺溶液一段时间后,得到酪胺基团修饰的第二水凝胶(式v),第二水 凝胶在浸泡了hrp溶液或者酪氨酸酶溶液后得到两性离子水凝胶,后者在关节腔内能够二次 酶交联以提高水凝胶的力学性能。
[0022][0023]
在本技术方案中,两性离子单体优选采用sbma、cbma或mpc。在一个或多个实施例 中,也可以采用其他的两性离子单体。
[0024]
在一个或多个实施例中,将透明质酸-对乙酰氨基酚前药配制成0.5~2wt%的第二溶液, 将两性离子单体、nhsma、光引发剂和交联剂混合得到单体溶液,再将第二溶液和单体溶液 等体积混合。
[0025]
在部分优选的实施例中,第一水凝胶在酪胺溶液中浸泡12~48小时,优选地,浸泡18~24 小时。在一个或多个实施例中,第二水凝胶在辣根过氧化氢酶溶液或者酪氨酸酶溶液浸泡 12~48小时,优选地,浸泡20~30小时。
[0026]
进一步地,所述nhsma与两性离子单体的摩尔比为1:10~1:20,所述两性离子单体的体 积摩尔浓度为1~4m。
[0027]
进一步地,所述前药的含量为0.5~2.0wt%。
[0028]
进一步地,所述交联剂为pegda,所述光引发剂为i2959。
[0029]
进一步地,所述前药的制备包括以下步骤:
[0030]
将透明质酸钠溶解后加入4-二甲氨基吡啶和n,n
′‑
二环己基碳酰二亚胺反应一段时间后, 向反应液中加入对乙酰氨基酚继续反应,反应结束后透析、冻干得到所述前药。
[0031]
本技术方案中,将透明质酸钠(ha)加入二甲亚砜和水的混合溶剂中,二甲亚砜和水的 体积比为1∶1,在60℃下搅拌至完全溶解。之后,向透明质酸钠溶液中加入4-二甲氨基吡啶 (dmap)和n,n
′‑
二环己基碳酰二亚胺(dcc),继续反应2~4小时,随后加入对乙酰氨基 酚(pa)继续反应18~24小时。反应完成后,将反应液倒入透析袋中,透析袋的截留分子量 优选为3.5kda,透析后通过冻干得到前药产物。
[0032]
在部分优选的实施例中,所述透明质酸与对乙酰氨基酚的质量比为1.2~5.2∶1。
[0033]
进一步地,所述第一水凝胶具有式ii~式iv所示的结构:
[0034][0035]
进一步地,所述第二水凝胶具有式v~式vii所示的结构,即酪胺基团修饰后:
[0036][0037]
本发明的又一个目的在于提供一种基于前述任一制备方法得到的两性离子水凝胶,该两 性离子不仅能够在关节环境内缓慢地释放透明质酸-对乙酰氨基酚前药,通过透明质酸的靶向 作用实现炎症的靶向治疗,而且其在过氧化氢过表达的骨关节炎环境中能够进一步提高自身 的力学性能,避免关节环境中的剪切力对两性离子水凝胶植入体的损伤。
[0038]
本发明的又一个目的在于提供前述任一种两性离子水凝胶在骨关节腔环境中的应用,具 体地,所述两性离子水凝胶用于在骨关节环境中缓慢释放式i所示的前药,且所述两性离子 水凝胶在过氧化氢过表达的骨关节环境中力学性能增强。
[0039]
本发明与现有技术相比,具有如下的优点和有益效果:
[0040]
1、本发明提供的透明质酸-对乙酰氨基酚前药能够通过紫外光交联接枝到两性离子水凝 胶上,可以在关节环境中持续缓慢释放,相较于传统的小分子、游离状的透明质酸、对乙酰 氨基酚对骨关节炎的治疗效果更好,并且,该前药接枝在两性离子水凝胶上后不会影响两性 离子水凝胶、透明质酸、对乙酰氨基酚对关节的润滑和抗炎功能;
[0041]
2、本发明提供的两性离子水凝胶不仅能够在关节环境内缓慢地释放透明质酸-对
乙酰氨 基酚前药,通过透明质酸的靶向作用实现炎症的靶向治疗,而且其在过氧化氢过表达的骨关 节炎环境中能够进一步提高自身的力学性能,避免关节环境中的剪切力对两性离子水凝胶植 入体的损伤;
[0042]
3、本发明提供的制备方法易于实施,不需要特殊的设备,易于实现批量生产与应用推广。
附图说明
[0043]
此处所说明的附图用来提供对本发明实施例的进一步理解,构成本技术的一部分,并不 构成对本发明实施例的限定。在附图中:
[0044]
图1为本发明具体实施例中透明质酸-对乙酰氨基酚前药的核磁共振氢谱(1hnmr)图;
[0045]
图2为本发明具体实施例中透明质酸-对乙酰氨基酚前药的紫外吸收光谱;
[0046]
图3为本发明具体实施例中两性离子水凝胶制备方法的流程框图;
[0047]
图4为本发明具体实施例中磺酸甜菜碱基水凝胶的红外吸收光谱;
[0048]
图5为本发明具体实施例中磺酸甜菜碱基水凝胶的扫描电镜图,50μm;
[0049]
图6为本发明具体实施例中羧酸甜菜碱基水凝胶的红外吸收光谱;
[0050]
图7为本发明具体实施例中磷酸胆碱基水凝胶的红外吸收光谱;
[0051]
图8为本发明具体实施例中三种两性离子水凝胶的抗污性能结果,四条曲线从上至下依 次为control、ezh、gelatin和水;
[0052]
图9为本发明具体实施例中透明质酸-对乙酰氨基酚前药(ha-pa)和两性离子水凝胶 (hydrogel)的细胞毒性;
[0053]
图10为本发明具体实施例中透明质酸-对乙酰氨基酚前药和两性离子水凝胶的活/死染色 图片,图中箭头所指为死细胞;
[0054]
图11为本发明具体实施例中透明质酸-对乙酰氨基酚前药和两性离子水凝胶对细胞内羟 基自由基的效果对比图;
[0055]
图12为本发明具体实施例中采用lps诱导细胞、用透明质酸-对乙酰氨基酚前药继续培 养细胞(lps+ha-pa)和两性离子水凝胶继续培养细胞(lps+hydrogel)对细胞一氧化氮产 生的抑制效果;
[0056]
图13示出了本发明具体实施例中未被酪胺修饰的第一水凝胶(zh)(磺酸甜菜碱基)、 酪氨酸修饰后的两性离子水凝胶(tzh)、以及过氧化氢过表达环境下(0.1~1.0mm)的酶 催化交联后两性离子水凝胶(ezh)的压缩力学性能,其中,图13(a)为应力-应变曲线, 图13(b)为压缩模量;
[0057]
图14示出了本发明具体实施例中未被酪胺修饰的第一水凝胶(zh)(磺酸甜菜碱基)、 酪氨酸修饰后的两性离子水凝胶(tzh)、以及过氧化氢过表达环境下(0.1~1.0mm)的酶 催化交联后两性离子水凝胶(ezh)的(a)储能模量、(b)损耗模量、(c)粘性系数、(d) 弹性系数。
具体实施方式
[0058]
为使本发明的目的、技术方案和优点更加清楚明白,下面结合实施例和附图,对本
发明 作进一步的详细说明,本发明的示意性实施方式及其说明仅用于解释本发明,并不作为对本 发明的限定。
[0059]
本发明所有原料,对其来源没有特别限制,在市场上购买的或按照本领域技术人员熟知 的常规方法即可制备。本发明所有原料,对其纯度没有特别限制,本发明优选采用分析纯或 水凝胶领域常规的纯度要求。本发明所有原料,其牌号和简称均属于本领域常规牌号和简称, 每个牌号和简称在其相关用途的领域内均是清楚明确的,本领域技术人员根据牌号、简称以 及相应的用途,能够从市售中购买得到或者通过常规方法制备得到。
[0060]
本发明对所述取代基的表达方式没有特别限制,均采用本领域技术人员熟知的表达方式, 本领域技术人员基于常识,可根据其表达方式正确理解其含义。
[0061]
实施例1~3:透明质酸-对乙酰氨基酚(ha-pa)前药的制备
[0062]
ha-pa前药的合成路线为:
[0063][0064]
实施例1:
[0065]
将透明质酸钠0.4836g加入二甲亚砜和水的混合溶剂中并在60℃下搅拌至完全溶解,其 中,二甲亚砜和水的体积比为1∶1。加入0.0250g 4-二甲氨基吡啶和0.0625g n,n
′‑
二环己基碳 酰二亚胺,继续反应3h;然后加入0.0940g对乙酰氨基酚,继续反应24h。
[0066]
反应完成后将反应液倒入透析袋中,透析袋的截留分子量为3.5kda,透析后通过冻干得 到透明质酸-对乙酰氨基酚前药。
[0067]
实施例2:
[0068]
将透明质酸钠0.4836g加入二甲亚砜和水的混合溶剂中并在65℃下搅拌至完全溶解,其 中,二甲亚砜和水的体积比为1∶1。加入0.0250g 4-二甲氨基吡啶和0.0625g n,n
′‑
二环己基碳 酰二亚胺,继续反应4h;然后加入0.1874g对乙酰氨基酚,继续反应24h。
[0069]
反应完成后将反应液倒入透析袋中,透析袋的截留分子量为3.5kda,透析后通过冻干得 到透明质酸-对乙酰氨基酚前药。试验结果如图1和图2所示。图1中,8.01ppm和6.89ppm 代表对乙酰氨基酚中苯环上的氢,药物中的-ch3与透明质酸自身的-ch3化学位移产生重合。 在图2中,对比未修饰的ha,ha-pa在280nm处有明显的紫外吸收峰,说明药物已经偶联 在ha分子链上。
[0070]
实施例3:
[0071]
将透明质酸钠0.4836g加入二甲亚砜和水的混合溶剂中并在60℃下搅拌至完全溶解,其 中,二甲亚砜和水的体积比为1∶1。加入0.0250g 4-二甲氨基吡啶和0.0625g n,n
′‑
二环己基碳 酰二亚胺,继续反应3h;然后加入0.3758g对乙酰氨基酚,继续反应22h。
[0072]
反应完成后将反应液倒入透析袋中,透析袋的截留分子量为3.5kda,透析后通过冻干得 到透明质酸-对乙酰氨基酚前药。
[0073]
实施例4~6:两性离子水凝胶的制备
[0074]
实施例4:
[0075][0076]
将实施例2制得的透明质酸-对乙酰氨基酚前药配置成浓度为0.5~2wt%的溶液,将磺酸 甜菜碱甲基丙烯酸酯(sbma)、nhsma、pegda、i2959按照表1的配方比例进行混合后 加入去离子水至10ml得到单体溶液,混合所述单体溶液和透明质酸-对乙酰氨基酚前药溶液 后紫外光照成胶得到具有式ii的结构第一水凝胶。试验结果如图4所示,1183cm-1
和1038cm-1
两处特征吸收峰说明水凝胶中磺酸甜菜碱结构的存在,3447cm-1
处特征吸收峰说明水凝胶中 酰胺结构,主要来自于ha-pa以及nhs基团。
[0077]
表1 ha-pa&psb水凝胶各组分比例
[0078][0079][0080]
将第一水凝胶浸泡于酪胺溶液(1m)中24小时,然后浸泡于去离子水中24小时以除去 未反应的酪胺分子,得到具有式v结构的第二水凝胶。试验结果如图4所示,酪氨酸修饰后 形成大量酰胺键,有明显的酰胺i带(1615cm-1
)和酰胺ii带(1520cm-1
)吸收峰,且整个 体系中的苯环结构增加,1598cm-1
处苯环的吸收峰也明显增强,3098cm-1
处出现的吸收峰属 于酪氨酸中的羟基。
[0081]
将第二水凝胶浸泡于辣根过氧化氢酶(hrp)溶液(1mm)中24小时,得到两性离子水 凝胶1。试验结果如图4所示,随着交联过程中酚羟基的消耗以及酶的引入,3098cm-1
处吸 收峰的相对强度减弱。水凝胶的微观结构如扫描电镜图(图5)所示,呈现多孔结构。
[0082]
实施例5:
[0083][0084]
将实施例2制得的透明质酸-对乙酰氨基酚前药配置成浓度为0.5~2wt%的溶液,将羧酸 甜菜碱甲基丙烯酸酯(cbma)、nhsma、pegda、i2959按照表2的配方进行混合后加入 去离子水至10ml得到单体溶液,混合所述单体溶液和透明质酸-对乙酰氨基酚前药溶液后紫 外光照成胶得到具有式iii结构的第一水凝胶。试验结果如图6所示,1143cm-1
属于c-o结 构的吸收峰,主要来自于羧酸甜菜碱,3447cm-1
处为酰胺结构,主要来自于ha-pa和nhs。
[0085]
表2 ha-pa&pcb水凝胶各组分比例
[0086][0087]
将第一水凝胶浸泡于酪胺溶液(1m)中24小时,然后浸泡于去离子水中24小时以除去 未反应的酪胺分子,得到具有式vi结构的第二水凝胶。试验结果如图6所示,酪氨酸修饰后 形成大量酰胺键,有明显的酰胺i带(1616cm-1
)和酰胺ii带(1519cm-1
)吸收峰,且整个 体系中的苯环结构增加,1590cm-1
处苯环的吸收峰也明显增强,3104cm-1
处出现的吸收峰属 于酪氨酸中的羟基。
[0088]
将第二水凝胶浸泡于酪氨酸酶溶液(1mm)中24小时,得到两性离子水凝胶2。试验结 果如图6所示,随着交联过程中酚羟基的消耗以及酶的引入,3104cm-1
处吸收峰的相对强度 减弱。
[0089]
实施例6:
[0090][0091]
将实施例1制得的透明质酸-对乙酰氨基酚前药配置成浓度为0.5~2wt%的溶液,将磷酸 胆碱甲基丙烯酸酯(mpc)、nhsma、pegda、i2959按照表3的配方进行混合后加入去离 子水至10ml得到单体溶液,混合所述单体溶液和透明质酸-对乙酰氨基酚前药溶液后紫外光 照成胶得到具有式iv结构的第一水凝胶。试验结果如图7所示,962cm-1
和1243cm-1
两处 特征吸收峰分别是磷酸胆碱结构中的p-o-c和o=p-o-,3447cm-1
处特征吸收峰说明水凝胶 中酰胺结构,主要来自于ha-pa以及nhs基团。
[0092]
表3 ha-pa&pc水凝胶各组分比例
[0093]
[0094][0095]
将第一水凝胶浸泡于酪胺溶液(1m)中24小时,然后浸泡于去离子水中24小时以除去 未反应的酪胺分子,得到具有式vii结构的第二水凝胶。试验结果如图7所示,酪氨酸修饰 后形成大量酰胺键,有明显的酰胺i带(1616cm-1
)和酰胺ii带(1519cm-1
)吸收峰,且整 个体系中的苯环结构增加,1593cm-1
处苯环的吸收峰也明显增强,3108cm-1
处出现的吸收峰 属于酪氨酸中的羟基。
[0096]
将第二水凝胶浸泡于辣根过氧化氢酶(hrp)溶液(1mm)中24小时,得到两性离子水 凝胶3。试验结果如图7所示,随着交联过程中酚羟基的消耗以及酶的引入,3108cm-1
处吸 收峰的相对强度减弱。
[0097]
实施例7~12:两性离子水凝胶验证实验
[0098]
实施例7:
[0099]
本实施例中,选用实施例4中所制备的两性离子水凝胶1,通过荧光分光光度计与紫外灯 检测水凝胶的“抗污”性能。其具体检测步骤如下:
[0100]
(1)制备fitc标记的牛血清蛋白(fitc-bsa)
[0101]
首先,将10mg牛血清蛋白(bsa)溶解于5ml磷酸盐缓冲液(pbs,ph=7.2-7.4)中, 然后将fitc溶解于200μl dmso中,冰浴条件下将fitc溶液缓慢滴加到bsa溶液中,避光反 应24小时。待反应结束后,避光透析(mwco 1,000)3天,冻干后保存在-20℃环境下。
[0102]
(2)荧光分光光度计测试
[0103]
将水凝胶(500mg)与fitc-bsa溶液(1mg/ml)在37℃水浴中共培养24小时,然后将 水凝胶取出并洗涤,用荧光分光光度计检测剩余溶液的荧光强度与初始配置的fitc-bsa溶液 的荧光强度的差异。参数设置:激发波长为488nm,扫描范围是500-600nm。试验结果如图8 所示,control代表fitc-bsa溶液,ezh代表磺酸甜菜碱基两性离子水凝胶,gelatin代表明胶 水凝胶。明胶水凝胶对于蛋白没有优异的抗污性能,因此,fitc-bsa被大量吸附,剩余溶液 的荧光强度明显降低;ezh具有抗污性能,因此fitc-bsa只有少量吸附在水凝胶中,荧光强 度没有明显降低。
[0104]
实施例8:
[0105]
本实施例中,选用实施例2中制备的对ha-pa前药和实施例4中制备的两性离子水凝胶1, 通过cck-8法与活/死染色检测水两性离子水凝胶的细胞相容性。其具体步骤如下:
[0106]
cck-8法:将生长至对数生长期的大鼠软骨细胞按照10000/孔的密度接种于96孔板中, 继续培养24小时直至细胞完全贴壁。随后,使用含有透明质酸-对乙酰氨基酚前药以及两性离 子水凝胶浸提液的培养基继续培养细胞24小时。最后,加入cck-8试剂,继续培养2小时,测 量450nm处的吸光度。试验结果如图9所示,ha-pa以及所制备的水凝胶的浸提液
与细胞共培 养后,细胞存活率均超过90%,说明所制备的ha-pa前药以及水凝胶具有良好的细胞相容性。
[0107]
活/死染色:细胞培养过程与cck-8法一致,待与含有透明质酸-对乙酰氨基酚前药以及两 性离子水凝胶浸提液的培养基继续培养细胞24小时后,将培养基更换为活/死染色工作液体 (二乙酸荧光素:50μg/ml;碘化丙啶:10μg/ml),继续孵育15分钟后,采用荧光显微镜 拍照。试验结果如图10所示,与对照组类似,与ha-pa以及所制备的水凝胶的浸提液共培养 后的细胞,没有出现明显的死亡情况,大量的活细胞存在说明所制备的ha-pa前药以及水凝 胶对细胞的生长没有不利影响。
[0108]
实施例9
[0109]
本实施例中,选用实施例2中制备的对ha-pa前药和实施例4中制备的两性离子水凝胶1, 采用dcfh-da荧光探针检测细胞内羟基自由基表达情况,其具体步骤如下:
[0110]
将生长至对数生长期的大鼠软骨细胞按照10000/孔的密度接种于96孔板中,继续培养24 小时直至细胞完全贴壁。接下来,采用脂多糖(lps,1μg/ml)诱导细胞24小时。随后,使 用含有透明质酸-对乙酰氨基酚前药以及两性离子水凝胶浸提液的培养基继续培养细胞24小 时。最后,将dcfh-da探针装载进细胞,采用荧光显微镜检测细胞内羟基自由基的表达情况。 试验结果如图11所示,lps诱导后,出现明显的荧光信号,说明细胞内产生了羟基自由基, 当与ha-pa以及所制备的水凝胶的浸提液共培养后,细胞内的荧光信号减弱,说明ha-pa以 及所制备的水凝胶对于羟基自由基具有清除作用。
[0111]
实施例10
[0112]
本实施例中,选用实施例2中制备的对ha-pa前药和实施例4中制备的两性离子水凝胶1, 采用一氧化氮检测试剂盒检测细胞外一氧化氮的表达与累积。其具体步骤如下:
[0113]
将大鼠软骨细胞培养至对数生长期,然后将细胞接种于96孔板中,接种密度为10000/孔。 待细胞完全贴壁后,加入lps(1μg/ml)诱导细胞24小时,然后采用含有透明质酸-对乙酰氨 基酚前药以及两性离子水凝胶浸提液的培养基继续培养细胞24小时,将与细胞共培养的培养 基收集并采用一氧化氮检测试剂盒测试细胞外一氧化氮的表达与累积量。试验结果如图12所 示,lps诱导后的细胞的培养基中累积的no比对照组更高,说明细胞的炎症反应已经激活; 当细胞与ha-pa以及所制备的水凝胶的浸提液共培养后,培养基中的no降低,说明ha-pa 以及所制备的水凝胶可以抑制no的表达进而减缓炎症反应,且水凝胶组的no表达与对照组 相近。
[0114]
实施例11
[0115]
本实施例中,选用实施例2中制备的对ha-pa前药和实施例4中制备的两性离子水凝胶1, 通过万能力学试验机测试水凝胶的力学性能。其测试步骤如下:
[0116]
将所制备的水凝胶裁剪成为“底10mm
×
高8mm”的圆柱体。然后按照2mm/min的速度 进行压缩实验,比较水凝胶样品浸泡过过氧化氢前后的压缩力学性能差异。试验结果如图13 所示,zh代表两性离子水凝胶,tzh代表酪氨酸修饰后的两性离子水凝胶,ezh代表在辣根 过氧化氢酶/h2o2催化交联后的两性离子水凝胶;修饰酪氨酸后,水凝胶中存在部分疏水微区, tzh压缩模量提高;交联后,水凝胶网络中的交联点增加,ezh力学性能进一步提高。
[0117]
实施例12
[0118]
本实施例中,通过流变仪测试水凝胶的储能和损耗模量。其测试步骤如下:
[0119]
水凝胶制备的尺寸是:底10mm、高1mm的圆片。然后将水凝胶进行频率扫描和应变扫 描。频率扫描的参数设置为:应变1%,频率范围0.1hz-10hz。应变扫描的参数设置为:频 率1hz,应变范围0.1%-100%。试验结果如图14所示,zh代表两性离子水凝胶,tzh代表酪 氨酸修饰后的两性离子水凝胶,ezh代表在辣根过氧化氢酶/h2o2催化交联后的两性离子水凝 胶;三种水凝胶的储能模量都高于损耗模量,满足凝胶的性能。此外,三种水凝胶的损耗系 数大小排序依次是zh》tzh》ezh,说明酪氨酸的修饰以及辣根过氧化氢酶/h2o2催化后的二 次交联可以降低水凝胶的粘性,同样地,弹性系数的大小排序(zh》tzh》ezh)说明水凝胶 弹性的提高,ezh的弹性最好,接近于弹性体。
[0120]
本文中所使用的“第一”、“第二”等(例如第一溶液、第二溶液,第一水凝胶、第二水 凝胶等)只是为了描述清楚起见而对相应部件进行区别,不旨在限制任何次序或者强调重要 性等。在发明中使用的术语“连接”在不进行特别说明的情况下,可以是直接相连,也可以 使经由其他部件、基团间接相连。
[0121]
以上所述的具体实施方式,对本发明的目的、技术方案和有益效果进行了进一步详细说 明,所应理解的是,以上所述仅为本发明的具体实施方式而已,并不用于限定本发明的保护 范围,凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本 发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1