一种基于划窗映射的基因组测序数据快速注释方法和系统与流程
1.本发明属于生物信息技术领域,具体地,涉及一种基于划窗映射的基因组测序数据快速注释方法和系统。
背景技术:
2.下一代测序(next-generation sequencing,ngs)又称为高通量测序(high-throughput sequencing),是基于pcr和基因芯片发展而来的边合成边测序技术。高通量测序技术的特点主要有:测序读长短,通量高,准确度高。高通量测序相比一代测序大幅降低了成本,同时保持了较高准确性,并且大幅降低了测序时间,目前高通量测序已经在全组学得到广泛应用。比如:有参转录组测序,重测序,dna甲基化测序,m6a甲基化测序,单细胞测序等。
3.dna甲基化是表观遗传修饰的主要方式,能在不改变dna序列的前提下,改变遗传表现,在调控基因表达和染色质构象等方面发挥着重要作用。dna甲基化主要形成5-甲基胞嘧啶(5-mc)和少量的n6-甲基嘌呤(n6-ma)及7-甲基鸟嘌呤(7-mg)等。通常地,甲基化dna主要指5-甲基胞嘧啶(5mc)。哺乳动物细胞中甲基化主要发生在cg二核苷酸的胞嘧啶上,植物细胞中则存在很大比例的non-cg(chh、chg,h代表a、c、t)甲基化。5-甲基胞嘧啶(5-mc)由dna甲基转移酶(dna methyl-transferase,dnmt)催化s-腺苷甲硫氨酸(s-adenosylmethionine,sam)作为甲基供体,将胞嘧啶转变为5-甲基胞嘧啶(mc)。
4.全基因组甲基化测序(whole-genome bisulphite sequencing,wgbs)结合亚硫酸氢盐转化(bisulfite conversion)方法与二代测序技术,可在单碱基分辨率水平上高效地检测全基因组dna甲基化状态。亚硫酸氢盐处理可以使dna中未发生甲基化的胞嘧啶脱氨基转变成尿嘧啶,而甲基化的胞嘧啶保持不变;pcr扩增所需片段,则尿嘧啶全部转化成胸腺嘧啶。对pcr产物进行高通量测序,与参考序列比对,即可判断cpg/chg/chh位点是否发生甲基化。全基因组甲基化测序可全面、精确地检测全基因组dna甲基化状态,为更深入的表观遗传调控分析奠定基础。
5.基因启动子区cpg island通常为去甲基化状态,促进基因转录,而异常甲基化会导致转录的失活。一般而言,cpg island甲基化会导致基因沉默。dna甲基化在基因组印记方面有重要作用,在双等位基因中的一个发生高甲基化会导致单等位基因表达。
6.目前的生物信息软件对于dna甲基化测序数据在启动子区(promoter),外显子区(exonic),内含子区(intronic)和基因间区(intergenic)等基因结构区域的注释和cpg island区域的注释没有一致和快速的注释方法。
技术实现要素:
7.为了解决上述技术问题中的至少一个,本发明采取的技术方案如下:
8.本发明第一方面提供一种基于划窗映射的基因组测序数据快速注释方法,包括以下步骤:
9.s1,建立索引文件:
10.获得测序样本来源物种的功能组件区的起始位点和终止位点,对于每一个功能组件,利用公式(1)获得映射值:
[0011][0012]
其中,gi代表第i个位点的映射值,int表示取整数,si代表第i个位点数值,si代表第i个位点所属的划窗区间的起始位点,w代表划窗大小,w大小根据所述来源物种的所有功能组件区长度确定,
[0013]
由此获得所有功能组件区的起始位点和终止位点的映射值以及位于功能组件区内的窗口起点/终点的映射值,按下列格式构建索引文件:
[0014]
chr s e s e function
[0015]
其中,chr代表功能组件区所在染色体位置信息,s代表功能组件区起始位点或者位于功能组件区内的划窗起点的映射值,e代表功能组件区终止位点或者位于功能组件区内的划窗终点的映射值,s代表功能组件区起始位点,e代表功能组件区终止位点,function代表功能组件区的类别;
[0016]
s2,获得待注释位点的映射值:其位点数值为q,同样利用公式(1)获得待注释位点的映射值g;
[0017]
s3,将步骤s2获得的映射值g在所述索引文件的第2列和第3列进行搜索,若对于某一行j,g满足sj≤g≤ej,进一步判断q是否满足sj≤q≤ej,若满足,则所述待注释位点可注释位于第j行对应的功能组件区。
[0018]
在本发明的一些实施方案中,所述w的确定方法具体如下:
[0019]
(1)获得所述来源物种的所有功能组件区的长度;
[0020]
(2)获得所有功能组件区长度的代表值,根据代表值确定w值。
[0021]
在本发明的一些具体实施方案中,所述代表值选自中位数、众数、平均数、1/4分位数和3/4分位数中的一种,所述w值代表值。在本发明的另一些具体实施方案中,所述w值位于所述1/4分位数和3/4分位数之间。
[0022]
在这里,w值的获得是本发明意外发现可以使得处理后注释效率更高的选取方法,本领域技术人员也可以利用其他方式选取w值,只要不违背本发明的核心思想,都应视为落入本发明的保护范围。
[0023]
在本发明的一些实施方案中,所述来源物种为哺乳动物。优选地,所述来源物种为人。
[0024]
在本发明的一些实施方案中,所述基因测序数据是指dna甲基化测序数据。
[0025]
在本发明的一些实施方案中,所述功能组件区包括启动子区、外显子区、内含子区、promoter cgis、intragenic cgis、3'transcript cgis、intergenic cgis、重复区和mirna区。
[0026]
其中,promoter cgis、intragenic cgis、3'transcript cgis、intergenic cgis是根据cgi所属的基因位置进行定义的:
[0027]
promoter cgis-1000bp tss to+300bp tssintragenic cgis+300bp tss to+300bp tes
3'transcript cgis-300bp tes to+300bp tesintergenic cgis-300bp tes to-1000bp next gene's promoter
[0028]
本发明第二方面提供一种基于划窗映射的基因组测序数据快速注释系统,包括以下模块:
[0029]
索引库模块,用于存储索引文件,其中,所述索引文件的构建方法如下:
[0030]
获得测序样本来源物种的功能组件区的起始位点和终止位点,对于每一个功能组件,利用公式(1)获得映射值:
[0031][0032]
其中,gi代表第i个位点的映射值,int表示取整数,si代表第i个位点数值,si代表第i个位点所属的划窗区间的起始位点,w代表划窗大小,w大小根据所述来源物种的所有功能组件区长度确定,
[0033]
由此获得所有功能组件区的起始位点和终止位点的映射值以及位于功能组件区内的窗口起点/终点的映射值,按下列格式构建索引文件:
[0034]
chr s e s e function
[0035]
其中,chr代表功能组件区所在染色体位置信息,s代表功能组件区起始位点或者位于功能组件区内的划窗起点的映射值,e代表功能组件区终止位点或者位于功能组件区内的划窗终点的映射值,s代表功能组件区起始位点,e代表功能组件区终止位点,function代表功能组件区的类别,
[0036]
输入模块,用于接收测序数据,获得待注释位点,并利用公式(1)计算待注释位点的索引值,
[0037]
搜索模块,分别与输入模块和索引库模块连接,用于将输入模块获得的所述待注释位点的索引值在所述索引文件的第2列和第3列进行搜索,若对于某一行j,g满足sj≤g≤ej,进一步判断q是否满足sj≤q≤ej,若满足,则所述待注释位点可注释位于第j行对应的功能组件区,
[0038]
结果输出模块,用于将注释结果输出。
[0039]
在本发明的一些实施方案中,所述w的确定方法具体如下:
[0040]
(1)获得所述来源物种的所有功能组件区的长度;
[0041]
(2)获得所有功能组件区长度的代表值,根据代表值确定w值。
[0042]
在本发明的一些具体实施方案中,所述代表值选自中位数、众数、平均数、1/4分位数和3/4分位数中的一种,所述w值代表值。在本发明的另一些具体实施方案中,所述w值位于所述1/4分位数和3/4分位数之间。
[0043]
在本发明的一些实施方案中,所述来源物种为哺乳动物。优选地,所述来源物种为人。
[0044]
在本发明的一些实施方案中,所述基因测序数据是指dna甲基化测序数据。
[0045]
在本发明的一些实施方案中,所述功能组件区包括启动子区、外显子区、内含子区、promoter cgis、intragenic cgis、3'transcript cgis、intergenic cgis、重复区和mirna区。
[0046]
其中,promoter cgis、intragenic cgis、3'transcript cgis、intergenic cgis
是根据cgi所属的基因位置进行定义的:
[0047]
promoter cgis-1000bp tss to+300bp tssintragenic cgis+300bp tss to+300bp tes3'transcript cgis-300bp tes to+300bp tesintergenic cgis-300bp tes to-1000bp next gene's promoter
[0048]
本发明的有益效果
[0049]
相对于现有技术,本发明具有如下有益效果:
[0050]
利用本发明的方法和系统,通过对功能组件位置进行映射,方法简单易操作,并可以大幅提高搜索注释效率。以人类1号染色体为例,搜索效率可以提升497506倍。对于多个染色体及多个样本的注释,效果更加明显。
附图说明
[0051]
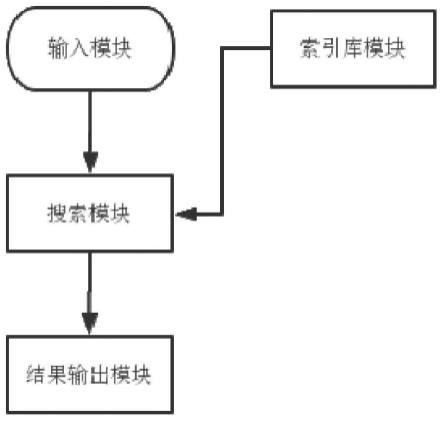
图1示出了cgi所属的基因位置。
[0052]
图2示出了一个位点(10540)位于一个启动子区([10300,13000])中的示意图。
[0053]
图3示出了本发明基于划窗映射的基因组测序数据快速注释系统示意图。。
具体实施方式
[0054]
除非另有说明、从上下文暗示或属于现有技术的惯例,否则本技术中所有的份数和百分比都基于重量,且所用的测试和表征方法都是与本技术的提交日期同步的。在适用的情况下,本技术中涉及的任何专利、专利申请或公开的内容全部结合于此作为参考,且其等价的同族专利也引入作为参考,特别这些文献所披露的关于本领域中的合成技术、产物和加工设计、聚合物、共聚单体、引发剂或催化剂等的定义。如果现有技术中披露的具体术语的定义与本技术中提供的任何定义不一致,则以本技术中提供的术语定义为准。
[0055]
本技术中的数字范围是近似值,因此除非另有说明,否则其可包括范围以外的数值。数值范围包括以1个单位增加的从下限值到上限值的所有数值,条件是在任意较低值与任意较高值之间存在至少2个单位的间隔。例如,如果记载的是100至1000,意味着明确列举了所有的单个数值,例如100,101,102等,以及所有的子范围,例如100到166,155到170,198到200等。对于包含小于1的数值或者包含大于1的分数(例如1.1,1.5等)的范围,则适当地将1个单位看作0.0001,0.001,0.01或者0.1。对于包含小于10(例如1到5)的个位数的范围,通常将1个单位看作0.1。这些仅仅是想要表达的内容的具体示例,并且所列举的最低值与最高值之间的数值的所有可能的组合都被认为清楚记载在本技术中。
[0056]
术语“包含”,“包括”,“具有”以及它们的派生词不排除任何其它的组分、步骤或过程的存在,且与这些其它的组分、步骤或过程是否在本技术中披露无关。为消除任何疑问,除非明确说明,否则本技术中所有使用术语“包含”,“包括”,或“具有”的组合物可以包含任何附加的添加剂、辅料或化合物。相反,出来对操作性能所必要的那些,术语“基本上由
……
组成”将任何其他组分、步骤或过程排除在任何该术语下文叙述的范围之外。术语“由
……
组成”不包括未具体描述或列出的任何组分、步骤或过程。除非明确说明,否则术语“或”指列出的单独成员或其任何组合。
[0057]
为了使本发明所解决的技术问题、技术方案及有益效果更加清楚明白,以下结合
实施例,对本发明进行进一步详细说明。
[0058]
实施例
[0059]
以下例子在此用于示范本发明的优选实施方案。本领域内的技术人员会明白,下述例子中披露的技术代表发明人发现的可以用于实施本发明的技术,因此可以视为实施本发明的优选方案。但是本领域内的技术人员根据本说明书应该明白,这里所公开的特定实施例可以做很多修改,仍然能得到相同的或者类似的结果,而非背离本发明的精神或范围。
[0060]
除非另有定义,所有在此使用的技术和科学的术语,和本发明所属领域内的技术人员所通常理解的意思相同,在此公开引用及他们引用的材料都将以引用的方式被并入。
[0061]
那些本领域内的技术人员将意识到或者通过常规试验就能了解许多这里所描述的发明的特定实施方案的许多等同技术。这些等同将被包含在权利要求书中。
[0062]
下述实施例中的实验方法,如无特殊说明,均为常规方法。下述实施例中所用的仪器设备,如无特殊说明,均为实验室常规仪器设备;下述实施例中所用的试验材料,如无特殊说明,均为自常规生化试剂商店购买得到的。
[0063]
实施例1基于dna甲基化测序进行功能组件的快速注释方法
[0064]
1.基因组功能组件分类与定义
[0065]
启动子是一个dna序列,蛋白质与该序列结合以从启动子下游的dna启动单个rna转录物的转录。rna转录物可以编码一种蛋白质(mrna),或者可以具有自身的功能(如trna或rrna)。启动子位于基因转录起始位点附近,位于dna上游(朝向有义链的5'区域)。启动子长度约为100-1000个碱基对,其序列高度依赖于基因和转录产物、类型或类别招募到该地点的rna聚合酶和生物种类。启动子区是rn a聚合酶的结合区,其结构直接关系到转录的效率。
[0066]
转录起始位点(tss)是指与新生rna链第一个核苷酸相对应dna链上的碱基,通常为一个嘌呤。常把起点前面,即5'末端的序列称为上游(upstream),而把其后面即3'末端的序列称为下游(downstream)。在描述碱基的位置时,一般用数字表示,tss起点为+1,下游方向依次为+2、+3
……
,上游方向依次为-1、-2、-3
……
。
[0067]
在本实施例中,发明人对于启动子区基于tss进行了统一定义,作为后续的dn a甲基化测序分析的统一注释标准,如表1所示:
[0068]
表1启动子区定义
[0069]
启动子区promoters-2,200至+500bp近端proximal(p)-200至+500bp中端intermediate(i)-200至-1,000bp远端distal(d)-1,000至-2,200bp
[0070]
启动子序列中的相对位置和cpg含量是启动子甲基化程度的重要影响因素,即o/e比值(observed-to-expected cpg ratio),根据该比值发明人将启动子区cpg的o/e值分为低、中、高(lcp,icp和hcp)三类。计算公式如下:
[0071][0072]
其中,num of cpg表示cpg数量,num of c表示序列中碱基c数量,num of g表示序列中碱基g数量,total number of nucleotides in the sequence表达序列中总碱基数
量。
[0073]
发明人根据cpg岛(cgi)所属的基因位置对cgi进行如表2所述定义:
[0074]
表2cgi定义
[0075][0076][0077]
cgi所属的基因位置定义如图1所示。
[0078]
2.利用位点平移法对序列所属功能组件区进行注释
[0079]
发明人根据dna甲基化测序的结果,将测序reads比对到基因组上,通常比对结果是以sam格式输出。sam格式中包括位点位置信息,pos:比对上的最左边的定位,即reads比对到基因组上的第一个碱基的位置情况。根据这个比对的位置情况,本领域人员需要快速注释到这个位点是处于基因的哪个功能区间(如启动子区、外显子区、内含子区),同时还需要确认是否处于cgi/cgi shores区域。
[0080]
在此,以人的基因组为例,人的1号染色体的长度为249250621个碱基,假设某几条reads比对到该染色体的第10540个碱基位置上,如何快速地通过位点搜索找到该位置(10540)是否属于启动子区成为本领域的一大难题。
[0081]
假设该位点是落在其中的一个基因的启动子区([10300,13000])内(如图2所示),如果按照单个位点平移的方法来进行遍历搜索注释,那么将循环10540次(从第1个碱基开始遍历)才能找到这个位点位于基因组的启动子区,该方法执行起来效率非常低。
[0082]
3.划窗映射法对序列所属功能组件区进行注释
[0083]
为了提高功能注释的搜索效率,发明人创建了一种快速注释的划窗映射搜索方法,详细过程如下:
[0084]
(1)首先,发明人采用如下公式来进行基因组的功能组件的映射创建:
[0085][0086]
其中,gi代表第i个位点的映射值,int表示取整数,si代表第i个位点数值,si代表第i个位点所属的划窗(window slide)区间的起始位点,w代表划窗大小。
[0087]
其中,w的大小根据来源物种的所有功能元件区的长度来确定。具体地:
[0088]
(1)获得来源物种的所有功能组件区的长度;
[0089]
(2)获得所有功能组件区长度的代表值,根据代表值确定w值。
[0090]
以人为例,代表值为1/4分位数和3/4分位数,分别是150和2000。在1/4分位数和3/4分位数之间选取w值,经过选取,w=500。
[0091]
以上面的启动子区[10300,13000]为例,该启动子区起始位点(10300),si=10300,由划窗法可知,si=10000,gi=10300/500=20。
[0092]
采用划窗法可知,该启动子区位于第一个划窗的终止位点是10500(10000+500),相应的索引值为:gj=10500/500=21。以此类推,可得到该启动子区内其他划窗起点/终点的映射值分别为:11000/500=22、11500/500=23、12000/500=24、12500/50=25、13000/
500=26,该启动子的终止位点刚好也是一个划窗的终止位点。
[0093]
发明人以该方法对该启动子区创建如下的索引进行存储,格式如下:
[0094]
chrsesefunctionchr120211030013000promoterchr121221030013000promoterchr122231030013000promoterchr123241030013000promoterchr125261030013000promoter
[0095]
其中,chr代表功能组件区所在染色体位置信息,s代表功能组件区起始位点或者位于功能组件区内的划窗起点的映射值,e代表功能组件区终止位点或者位于功能组件区内的划窗终点的映射值,s代表功能组件区起始位点,e代表功能组件区终止位点,function代表功能组件区的类别。功能组件区的类别包括但不限于:启动子区(promoter)、外显子区(exonic)、内含子区(intronic)、promoter cgis、intragenic cgis、3'transcript cgis、intergenic cgis、重复区(repeat region)和mirna区。
[0096]
对已知的所有功能组件均进行类似的索引转换得到索引文件。
[0097]
通过该方法,发明人对于原来的位点进行了数据压缩,[20,26]该区间的对应的原始位点为:[10000,13000],即用7个位点存储了原来3000个位点,搜索效率可以提升429倍,对于数据宠大的基因组数据,效果十分明显。
[0098]
注释时,只需要进行注释的位点,也进行相应索引值的转化后即可进行注释:
[0099]gk
=k/500
[0100]
其中,gk代表位点k的映射值,sk代表位点k的位点数值。
[0101]
搜索的时候,根据gk到索引文件里去搜索第二列(s)和第二列(e)数据,同时,发明人根据查询的位点查找是否落在第三列和第四列区间内,如果s≤gk≤e,并且s≤sk≤e那么位点k可以注释成相应的功能组件。
[0102]
例如对于上述10540这个位点,利用上述方法进行相应索引值的转化,转化后的数值如下:
[0103]gk
=10540/500=21
[0104]
该位点满足20≤21≤21,并且10300≤10540≤13000,因此位点k被注释为启动子。
[0105]
实施例2基于dna甲基化测序进行功能组件的快速注释方法的应用
[0106]
本实施例以1号染色体为例,1号染色体的序列总共有249250621个碱基,如果按传统的位点遍历的方法,需要最多遍历249250621次才能找到注释的位点。而按照实施例1的划窗映射搜索法,w=500,最多将分为498502个索引值。搜索时最多501次搜索即可以找到该位点,搜索效率提升了497506倍。
[0107]
具体的效率提升比较如表3:
[0108]
表3效率比较
[0109]
[0110]
由此可见,利用实施例1的划窗映射搜索法,可以大幅提升搜索性能。
[0111]
实施例3基于划窗映射的基因组测序数据快速注释系统
[0112]
如图3所示,本实施例提供一种系统,来实现上述快速注释方法,该系统包括:
[0113]
索引库模块,用于存储上述构建的索引文件
[0114]
输入模块,用于接收测序数据,获得待注释位点q,并利用上述公式计算待注释位点q的索引值g,
[0115]
搜索模块,分别与输入模块和索引库模块连接,用于将输入模块获得的所述待注释位点的索引值在所述索引文件的第2列和第3列进行搜索,若对于某一行j,g满足sj≤g≤ej,进一步判断q是否满足sj≤q≤ej,若满足,则所述待注释位点可注释位于第j行对应的功能组件区,
[0116]
结果输出模块,用于将注释结果输出。
[0117]
在本发明提及的所有文献都在本技术中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本技术所附权利要求书所限定的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1