一种用于微乳制剂的油相调配方法及其应用与流程
1.本发明属于制药领域,具体涉及一种油相调配方法,其根据油相的理化特性以及表面活性剂的种类,在推导的公式指导下添加特定比例的游离脂肪酸或甘油酸酯,调控微乳系统的粒径、电位、澄明度、稳定性等核心性能,可以用于提高微乳体系增溶难溶性药物的能力。
背景技术:
2.微乳作为新型药物制剂的一种,由乳化剂、助乳化剂、油相在水相中自组装形成,可以显著增溶难溶性药物,在以药物增溶为导向的剂型开发中拥有广阔的应用前景。此外,作为一种热力学稳定体系,微乳及其载药系统拥有比其他新型制剂更优的稳定性,近年来逐渐吸引了药物研发人员的重视。然而,在微乳制备过程中,微乳的粒径、电位、澄明度、稳定性、包封率、载药量等核心特征与所采用的辅料类型和各辅料间配比密切相关。在特定的辅料组成和一定的配比范围内,药学研发人员对微乳的制备工艺以及微乳药物增溶有调控能力。但当辅料种类更换、处方配比更改后,需要重新建立处方工艺优化路径,工作量大、程序繁琐、花费多。因此,理清哪些核心要素能够决定微乳制备和理化性质的核心数据是至关重要的。
3.油酸等游离脂肪酸主要来源于自然界,以甘油酯的形式存在于动植物油脂中。将油酸含量高的油脂经过皂化、酸化分离,即可得到油酸。从化学结构上看,具有游离羧基和长达17个碳原子的单烯烃链,又称十八碳烯酸;羧基具有亲水性,而单烯烃链具有疏水性,因此油酸也常被用作表面活性剂。包括微乳在内的纳米粒子由于纳米尺寸效应,比表面积以及粒子间作用能较大,这种特殊的表面结构很容易形成团聚体。要稳定纳米粒子在液体中的分散体系,主要通过减少吸引力、增加排斥力、控制极性基团暴露、调整亲水亲油基团平衡等方式来抑制纳米粒子形成聚块或絮凝。
4.微乳所用到的表面活性剂又称乳化剂,主要作用是乳化、分散、增溶难溶性成分,使多组分在无需外力作用下自发形成均相体系。它主要通过相似相容原理完成疏水端的内卷和亲水端的外展,以此形成一个斥力与吸附力平衡,产生较高的能垒来预防和抵抗粒子聚结,保持粒子的均相稳定。但在众多表面活性剂中,表面活性剂与油相的优化配比需要大量实验去筛选和验证,一旦更换了表面活性剂或者油相的种类,摸索最优的处方工艺工作量很大,耗时耗力。譬如申请人团队前期的发明专利(cn201510567457.5)采用薏苡仁油为油相,rh40为表面活性剂,peg400为表面活性剂,以100:(50~75):(15~25)制备的微乳符合粒径小、体系稳定,溶液澄明的特点。但薏苡仁油的理化特性受到薏苡仁药材来源的决定,不同产地的薏苡仁药材提取出的薏苡仁油酸值、碘值、蛋白含量会有差异,因此在制备薏苡仁油微乳的过程中,一旦产地出现变更,需要大量实验去调整油-表面活性剂-助表面活性剂的比例和工艺。同样,申请人前期的发明专利(cn201410149596.1;cn201410149584.9)中采用的其它来源油相,如蓖麻油、橄榄油、白术油、鸦胆子油等,油/表面活性剂/助乳化剂以40:(10~35):(10~20)比例可以制备出粒径小、体系稳定,溶液澄明
的微乳,但一旦天然来源的油相更换批次或提取来源,上述比例可能偏离出最佳的制备工艺范围。
技术实现要素:
5.本发明的目的是提供一种油相调配方法,可以使得在采用不同品相和理化性质的油相与乳化剂搭配制备微乳系统时,均可以通过酸值或者游离脂肪酸比例的调整快速制得符合特定要求(如粒子尺寸、澄明度、稳定性)的微乳体系。并且这种微乳对难溶性药物的增溶能力相比现有技术得到进一步的提高,可以用于微乳相关的新剂型开发与应用。
6.为实现上述目的,本发明采用如下的技术方案:
7.一种用于微乳制剂的油相调配方法,包括如下步骤:
8.(1)选定微乳制剂的油相和表面活性剂,并测出所述油相的酸值ah、所述表面活性剂的hlb值;
9.其中,
10.所述油相选自薏苡仁油、薄荷油、防风挥发油、姜黄挥发油、广藿香挥发油或鱼腥草挥发油;
11.所述表面活性剂选自聚山梨酯(20、40、60)、聚氧乙烯(12、20)十六十八烷基醚、十六十八烷基硫酸钠、十六十八烷基醇、聚氧乙烯(40)氢化蓖麻油、聚氧乙烯(15)羟基硬脂酸、聚氧乙烯(35)蓖麻油、聚丙乙烯聚氧丙烯醚(124、188、338、407);
12.(2)选定油相平衡调节剂:
13.根据式1计算得到c%的值:
[0014][0015]
当c%为正值时,所述油相平衡调节剂选自甘油酸三油酸酯、甘油酸二油酸酯或甘油酸单油酸酯;
[0016]
当c%为负值时,所述油相平衡调节剂选自油酸、亚油酸、亚麻酸、月桂酸或辛酸;
[0017]
(3)确定油相平衡调节剂的用量:
[0018]
当所述微乳制剂的微乳粒径为30nm时,所述油相平衡调节剂在所述微乳制剂中的质量分数为|c%|;
[0019]
当所述微乳制剂的微乳粒径为其他粒径时,所述油相平衡调节剂在所述微乳制剂中的质量分数为a|c%|,其中a满足式2:
[0020]
d=-65.1 loga+30
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
式2;
[0021]
其中,d为所述微乳制剂的微乳粒径,单位是nm;
[0022]
(4)制备微乳制剂:
[0023]
将选定的油相、表面活性剂、油相平衡调节剂和助表面活性剂制成微乳制剂,其中油相和表面活性剂的质量比为1:4~4:1。
[0024]
优选的,步骤(4)包括如下步骤:
[0025]
(4-1)将所述油相和所述油相平衡调节剂混合得到匀质的调节油相后,向所述调节油相中加入所述表面活性剂混合均匀,之后加入助表面活性剂并混匀得到分散相溶液;
[0026]
(4-2)向所述分散相溶液中加入水混匀,得到所述微乳制剂。
[0027]
优选的,所述表面活性剂和所述助表面活性剂的质量比例为1:1~3:1。
[0028]
优选的,步骤(4-1)中混合均匀的方法为:搅拌2~6h。
[0029]
优选的,所述助表面活性剂为聚乙二醇、丙二醇、中链甘油三酯的一种。
[0030]
优选的,步骤(4-2)中缓慢滴加水并持续搅拌得到所述微乳制剂。
[0031]
优选的,步骤(4)还包括向所述油相中加入药物的步骤。
[0032]
优选的,所述药物为紫杉醇、长春碱、大麻二酚、紫草素、小檗碱、他克林、大麻二酚酸、脱氢大麻二酚、大麻艾尔松、大麻萜酚、大麻环酚、cbn大麻酚、二羟基大麻酚、四氢大麻酚、大麻环萜酚、黄芩苷、黄芪甲苷、芍药苷、白藜芦醇、连翘苷、木犀草苷、刺芒柄花素、葛根素、丹酚酸或丹皮酚中的一种或几种。
[0033]
本发明从油相角度出发,提出了一种独特的油相调配技术,根据酸值所反映出的油相中所含游离脂肪酸的比例,通过添加特定比例的游离脂肪酸或甘油酸酯,使得在油相更换或处方组成/配比更改后,可以快速将微乳的制备工艺及处方工艺调整到所需的最优区间,并能根据需要调控微乳及增溶药物。
[0034]
油酸结构式如下:
[0035][0036]
亚油酸结构式如下:
[0037][0038]
亚麻酸结构式如下:
[0039][0040]
月桂酸结构式如下:
[0041][0042]
甘油酸酯结构式如下:
[0043][0044]
以油酸为例,其为结构式中有一个烯烃双键(-ch=ch-)和一个羧基(-cooh)的长链不饱和脂肪酸,分子中的双键引起空间结构的弯曲,产生空间壁垒,阻碍了邻近链的集束。当所选取的油相需要用它来稳定纳米粒子时,它的疏水部分可以与表面活性剂中的疏水部分以相似相溶的原理被溶剂化,而暴露出的强极性基团可以校正油相的酸值、调整油相与表面活性剂的兼容度,让油相和表面活性剂疏水端压缩更紧密,从而形成更小的脂核,制备出的微乳粒子更小,溶液更清澈、结构更稳定、增溶难溶性药物的能力更强。同样的原理,当所选取的油相需要用甘油酸酯来稳定纳米粒子时,表明油相所含的极性基团过多,需要用甘油酸酯与表面活性剂中的疏水部分以相似相溶的原理被溶剂化,降低油相极性基团平均密度,同样达到校正油相的酸值、调整油相与表面活性剂的兼容度的目的。这种调配油相的效果也能让油相和表面活性剂疏水端压缩更紧密,形成脂核更小,微乳尺寸更小,溶液更澄明,提高药物增溶能力。
[0045]
申请人通过大量实验发现,根据酸值所反映出的油相中所含游离脂肪酸的比例,以及所采用的表面活性剂的hlb值,可以用游离脂肪酸或者甘油酸酯对油相进行调配处理,可以快速获得制备出粒径小、体系稳定,溶液澄明的微乳,并且各组分间的比例范围能够明显扩大。在增溶难溶性药物或者维持长时间储存稳定性方面也相较之前的处方有明显的改善。
[0046]
通过本发明的方法调配得到的微乳系统,与未经油相平衡调节的微乳相比,在微乳粒子尺寸、澄明度、稳定性、药物包封率、载药量、存储稳定性方面均具有显著的优势。加入油相平衡调节剂后,对微乳的核心性质调控行为产生了意想不到的优势,并且还能实现显著的药物增溶,有利于药学应用方面。
具体实施例
[0047]
实施例1
[0048]
(1)选定薏苡仁油(二等品)为油相,以聚氧乙烯(12)十六烷基十八烷基醚为表面活性剂,测得油相的酸值为ah=3.98,表面活性剂的hlb=16。
[0049]
(2)根据式1计算得到c%=-14.48%,为负值,据此选择油酸、亚油酸、亚麻酸或月桂酸作为油相平衡调节剂。
[0050]
(3)对于目标粒径60nm的乳液,根据式2计算得到a为0.34,则a|c%|=5.1%。
[0051]
对于目标粒径30nm的乳液,|c%|=2%。
[0052]
(4)将薏苡仁油和油相平衡调节剂于40℃下搅拌30min,完全匀质后,加入表面活性剂聚氧乙烯(12)十六烷基十八烷基醚,继续搅拌2~6h,再加入助表面活性剂丙二醇,使
得薏苡仁油、表面活性剂和助表面活性剂的质量比为1:2:0.7~2:1:0.4,之后将去离子水恒速、缓慢并定量地滴入,并不断匀速搅拌,直至出现透亮澄明的乳液。
[0053]
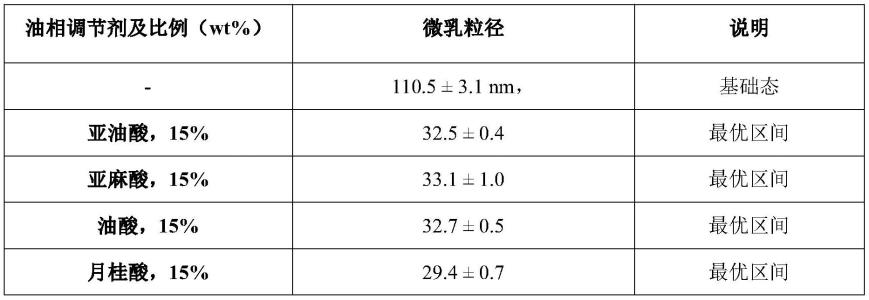
以不含油相平衡调节剂的相同体系以相同方法制备得到乳液作为基础态,测试所述基础态和不同比例和油相调节剂制得的乳液的微乳粒径,结果如表1所示:
[0054]
表1不同油相调节剂参与下薏苡仁油(二等品)的微乳粒径
[0055][0056][0057]
由表1可以看出,基础态下所制得的二等品薏苡仁油微乳粒径为110nm。当掺入15%油相负调节剂后,四种不同游离脂肪酸均能将微乳尺寸调整到30nm区间;当掺入5.2%的油相负调节剂后,微乳尺寸被调整至60nm区间。均与目标相符。
[0058]
作为对比,当加入正调节剂甘油酸二油酸酯后,微乳粒径出现不可控状态,分别增长至~320nm和~240nm。
[0059]
实施例2
[0060]
与实施例1步骤(1)、(2)类似,计算不同油相和表面活性剂配方下对应的c%,结果如表2所示:
[0061]
表2微乳粒径约30nm时不同处方下所需的油相平衡调节剂的量(wt%)
[0062][0063]
类似实施例1的步骤(3),计算不同油相和表面活性剂配方下对应的油相平衡调节剂的量(即ac%,并通过值的正负指导油相平衡调节剂的种类),结果如表3所示:
[0064]
表3微乳粒径约60nm时不同处方下所需的油相平衡调节剂的量(wt%)
[0065][0066]
实施例3油相调配方法对薏苡仁油制备微乳增溶cbd行为验证
[0067]
(1)~(3)与实施例1步骤(1)~(3)相同。
[0068]
(4)与实施例1步骤(4)的区别仅在于:薏苡仁油、表面活性剂和助表面活性剂的质量比为350:400:130,之后加入1~5wt%的cbd,之后将去离子水恒速、缓慢并定量地滴入,并不断匀速搅拌,直至得到均匀的薏苡仁油-大麻二酚微乳。
[0069]
以不含油相平衡调节剂的相同体系以相同方法制备得到乳液作为空白,测试空白和不同比例和油相调节剂制得的乳液的薏苡仁油-大麻二酚微乳的包封率,结果如表4所示:
[0070]
表4不同油相调节剂参与下薏苡仁油-大麻二酚微乳的包封率
[0071]
油相调节剂及用量(wt%)包封率(%)7天药物泄漏率(%)说明-78.1
±
2.4%66.9
±
5.2%空白亚油酸,15%89.4
±
1.9%36.3
±
1.4% 亚麻酸,15%93.7
±
1.1%34.7
±
0.2% 油酸,15%96.2
±
0.3%36.5
±
0.3% 月桂酸,15%92.2
±
1.0%33.5
±
1.9% 油酸,2%84.6
±
0.2%48.2
±
2.1% 亚油酸,2%88.1
±
1.1%46.9
±
2.7% 亚麻酸,2%85.2
±
0.2%42.2
±
0.4% 甘油酸二油酸酯,15%58.2
±
1.3%78.3
±
3.7% 甘油酸二油酸酯,5%64.5
±
0.4%77.9
±
2.1% [0072]
由表4可以看出,空白配方下薏苡仁油-大麻二酚微乳的包封率~80%,7天药物泄漏率为~67%。当加入15%负油相调节剂后,包封率可提升至90%以上,7天药物泄漏率也可以将低至~35%水平。当加入并不合适的正油相调节剂后,包封率降至~60%,7天药物泄漏率上升到~80%。
[0073]
可见,根据本发明的方法选择合适的油相调节剂和用量,不仅可以调整合适的微乳尺寸,还能改善所制得的微乳包封率和稳定性。并且,通过本发明所优化制得的薏苡仁油-大麻二酚微乳包封率和稳定性与粒子尺寸存在一定的负相关性。
[0074]
同样的增溶改善现象还出现在紫杉醇、长春碱、紫草素、他克林、大麻萜酚、大麻环酚、cbn大麻酚、二羟基大麻酚、四氢大麻酚、大麻环萜酚、黄芩苷、黄芪甲苷、芍药苷、白藜芦醇、连翘苷等难溶物研究实例中。
[0075]
实施例4
[0076]
(1)选定薄荷油为油相,以聚山梨酯40为表面活性剂,测得油相的酸值为ah=1.95,表面活性剂的hlb=13.6。
[0077]
(2)根据式1计算得到c%=19.72%,为正值,据此选择甘油酸三油酸酯、甘油酸二油酸酯或甘油酸单油酸酯作为油相平衡调节剂。
[0078]
(3)对于目标粒径60nm的乳液,根据式2计算得到a为0.34,则a|c%|=6.7%。
[0079]
对于目标粒径45nm的乳液,根据式2计算得到a为0.588,则a|c%|=11.8%。
[0080]
对于目标粒径30nm的乳液,|c%|=19.72%。
[0081]
(4)将薄荷油和油相平衡调节剂于40℃下搅拌30min,完全匀质后,加入表面活性剂,继续搅拌2~6h,再加入助表面活性剂聚乙二醇,使得薄荷油、表面活性剂和助表面活性剂的质量比为1:2:0.7~2:1:0.4,之后将去离子水恒速、缓慢并定量地滴入,并不断匀速搅拌,直至出现透亮澄明的乳液。
[0082]
以不含油相平衡调节剂的相同体系以相同方法制备得到乳液作为基础态,测试所述基础态和不同比例和油相调节剂制得的乳液的微乳粒径,结果如表5所示:
[0083]
表5不同油相调节剂参与下薄荷油微乳的粒径
[0084][0085][0086]
由表5可以看出,基础态下所制得的薄荷油微乳粒径为~135nm,当根据公式1的计算,掺入20%油相正调节剂后,三种不同游离脂肪酸均能将微乳尺寸调整到30nm区间;当掺入比例调整到11.8%后,微乳尺寸被调整至45nm区间。当掺入比例调整至6.7%后,微乳尺寸被调整至60nm区间。均与目标相符。
[0087]
作为对比,当加入20%负调节剂亚麻酸和油酸后,微乳粒径分别增长至~320nm和~240nm。负调节剂比例降至6%后,粒径又回落至~270nm。
[0088]
实施例5油相调配方法对薄荷油制备微乳增溶紫草素行为验证
[0089]
(1)~(3)与实施例4步骤(1)~(3)相同。
[0090]
(4)与实施例4步骤(4)的区别仅在于:薄荷油、表面活性剂和助表面活性剂的质量比为(250~450):(200~500):(70~160),之后加入0.5~6wt%的紫草素,之后将去离子水恒速、缓慢并定量地滴入,并不断匀速搅拌,直至得到均匀的薄荷油-紫草素微乳。
[0091]
以不含油相平衡调节剂的相同体系以相同方法制备得到乳液作为空白,测试空白和不同比例和油相调节剂制得的乳液的薄荷油-紫草素微乳的包封率,结果如表6所示:
[0092]
表6不同油相调节剂参与下薄荷油-紫草素微乳的包封率
[0093][0094][0095]
由表6可以看出,空白配方下薄荷油-紫草素微乳的包封率~70%,7天药物泄漏率为~74%。当加入20%正油相调节剂后,包封率可提升至~95%,7天药物泄漏率也可以将低至~40%水平。当正调节剂比例降低至11.8%后,包封率略有下降,7天药物泄漏率略有上升。当加入并不合适的负油相调节剂后,包封率迅速降至~50%以下,7天药物泄漏率上升到~80%以上。
[0096]
可见,根据本发明的方法选择合适的油相调节剂和用量,不仅可以调整合适的微乳尺寸,还能改善所制得的微乳包封率和稳定性。并且,通过本发明所优化制得的薄荷油-紫草素微乳包封率和稳定性与粒子尺寸存在一定的负相关性。
[0097]
同样的增溶改善现象还出现在紫杉醇、大麻二酚、长春碱、他克林、大麻萜酚、大麻环酚、cbn大麻酚、二羟基大麻酚、四氢大麻酚、大麻环萜酚、黄芩苷、黄芪甲苷、芍药苷、白藜芦醇、连翘苷等难溶物研究实例中。
[0098]
实施例6
[0099]
(1)选定防风挥发油为油相,以聚山梨酯40为表面活性剂,测得油相的酸值为ah=1.7,表面活性剂的hlb=13.6。
[0100]
(2)根据式1计算得到c%=15.40%,为正值,据此选择甘油酸三油酸酯、甘油酸二油酸酯或甘油酸单油酸酯作为油相平衡调节剂。
[0101]
(3)对于目标粒径60nm的乳液,根据式2计算得到a为0.34,则a|c%|=5.2%。
[0102]
对于目标粒径50nm的乳液,根据式2计算得到a为0.49,则a|c%|=7.8%。
[0103]
对于目标粒径30nm的乳液,|c%|=15.4%。
[0104]
(4)将防风挥发油和油相平衡调节剂于40℃下搅拌30min,完全匀质后,加入表面活性剂,继续搅拌2~6h,再加入助表面活性剂聚乙二醇,使得防风挥发油、表面活性剂和助表面活性剂的质量比为1:2:0.8~2:1:0.3,之后将去离子水恒速、缓慢并定量地滴入,并不断匀速搅拌,直至出现透亮澄明的乳液。
[0105]
以不含油相平衡调节剂的相同体系以相同方法制备得到乳液作为基础态,测试所述基础态和不同比例和油相调节剂制得的乳液的微乳粒径,结果如表7所示:
[0106]
表7不同油相调节剂参与下防风挥发油微乳的粒径
[0107][0108][0109]
由表7可以看出,掺入15.4%油相正调节剂后,三种不同游离脂肪酸均能将微乳尺寸调整到30nm区间;当掺入比例调整到7.8%后,微乳尺寸被调整至50nm区间。当掺入比例调整至5.2%后,微乳尺寸被调整至60nm区间。均与目标相符。
[0110]
作为对比,当加入20%负调节剂亚麻酸和油酸后,微乳粒径可增长至~400nm。负调节剂比例降至6%后,粒径又回落至~200nm。
[0111]
类似的,在以广藿香挥发油为油相的微乳构建方案中,根据表2设计的体系也有类似的结论。
[0112]
实施例7油相调配方法对防风挥发油制备微乳增溶黄芩苷的行为验证
[0113]
(1)~(3)与实施例6步骤(1)~(3)相同。
[0114]
(4)与实施例6步骤(4)的区别仅在于:防风挥发油、表面活性剂和助表面活性剂的质量比为(220~450):(200~440):(65~140),之后加入1~8wt%的黄芩苷,之后将去离子水恒速、缓慢并定量地滴入,并不断匀速搅拌,直至得到均匀的防风挥发油-黄芩苷微乳。
[0115]
以不含油相平衡调节剂的相同体系以相同方法制备得到乳液作为空白,测试空白和不同比例和油相调节剂制得的乳液的防风挥发油-黄芩苷微乳的包封率,结果如表8所示:
[0116]
表8不同油相调节剂参与下防风挥发油-黄芩苷微乳的包封率
[0117][0118][0119]
由表8可以看出,空白防风挥发油-黄芩苷微乳的黄芩苷包封率~60%,7天药物泄漏率为~80%。当加入15.4%正油相调节剂后,包封率可提升至~85%,7天药物泄漏率也可以将低至~30%水平。当正调节剂比例降低至7.8%后,包封率略有下降,7天药物泄漏率略有上升。当加入并不合适的负油相调节剂后,包封率迅速降至~33%以下,7天药物泄漏率上升到~90%以上。
[0120]
可见,根据本发明的方法选择合适的油相调节剂和用量,不仅可以调整合适的微乳尺寸,还能改善所制得的微乳包封率和稳定性。并且,通过本发明所优化制得的薄荷油-紫草素微乳包封率和稳定性与粒子尺寸存在一定的负相关性。
[0121]
同样的增溶改善现象还出现在紫杉醇、大麻二酚、紫草素、长春碱、他克林、大麻萜酚、大麻环酚、cbn大麻酚、二羟基大麻酚、四氢大麻酚、大麻环萜酚、黄芪甲苷、芍药苷、白藜芦醇、连翘苷等难溶物研究实例中。
[0122]
实施例8
[0123]
(1)选定姜黄挥发油为油相,以聚氧乙烯(12)十六烷基十八烷基醚为表面活性剂,测得油相的酸值为ah=4.0,表面活性剂的hlb=16。
[0124]
(2)根据式1计算得到c%=-14.54%,为负值,据此选择油酸、亚油酸、亚麻酸、月桂酸或辛酸作为油相平衡调节剂。
[0125]
(3)对于目标粒径60nm的乳液,根据式2计算得到a为0.34,则a|c%|=5%。
[0126]
对于目标粒径45nm的乳液,根据式2计算得到a为0.588,则a|c%|=8.8%。
[0127]
对于目标粒径30nm的乳液,|c%|=14.54%。
[0128]
(4)将姜黄挥发油和油相平衡调节剂于40℃下搅拌30min,完全匀质后,加入表面活性剂,继续搅拌2~6h,再加入助表面活性剂丙二醇,使得姜黄挥发油、表面活性剂和助表面活性剂的质量比为1:2:0.7~2:1:0.4,之后将去离子水恒速、缓慢并定量地滴入,并不断匀速搅拌,直至出现透亮澄明的乳液。
[0129]
以不含油相平衡调节剂的相同体系以相同方法制备得到乳液作为基础态,测试所述基础态和不同比例和油相调节剂制得的乳液的微乳粒径,结果如表9所示:
[0130]
表9不同油相调节剂参与下姜黄挥发油微乳的粒径
[0131][0132]
由表9可以看出,基础态下所制得的姜黄挥发油微乳粒径为146nm,当根据公式1的计算,掺入15%油相负调节剂后,四种不同游离脂肪酸均能将微乳尺寸调整到30nm区间;当比例调整到8.8%后,微乳尺寸被调整至45nm区间。当比例调整到5%后,微乳尺寸被调整至60nm区间。均与目标相符。
[0133]
作为对比,当加入正调节剂甘油酸三油酸酯后,微乳粒径出现大幅提高,分别增长至~415nm和~300nm。
[0134]
类似的,在以鱼腥草挥发油、茶树油为油相的微乳构建方案中,根据表2设计的体系也有类似的结论。
[0135]
实施例9油相调配方法对姜黄挥发油制备微乳增溶白藜芦醇的行为验证
[0136]
(1)~(3)与实施例8步骤(1)~(3)相同。
[0137]
(4)与实施例8步骤(4)的区别仅在于:姜黄挥发油、表面活性剂和助表面活性剂的质量比为350:400:130,之后加入0.7~6.5wt%的白藜芦醇,之后将去离子水恒速、缓慢并定量地滴入,并不断匀速搅拌,直至得到均匀的姜黄挥发油-白藜芦醇微乳。
[0138]
以不含油相平衡调节剂的相同体系以相同方法制备得到乳液作为空白,测试空白和不同比例和油相调节剂制得的乳液的姜黄挥发油-白藜芦醇微乳的包封率,结果如表10所示:
[0139]
表10不同油相调节剂参与下姜黄挥发油-白藜芦醇微乳的包封率
[0140]
油相调节剂及用量(wt%)包封率(%)7天药物泄漏率(%)说明-52.1
±
1.8%76.3
±
4.8%空白亚油酸,15%82.4
±
1.9%44.2
±
0.7% 亚麻酸,15%83.5
±
1.0%48.2
±
0.9% 油酸,15%84.2
±
0.7%46.4
±
0.5% 月桂酸,15%90.2
±
1.4%40.2
±
0.9% 亚麻酸,8.8%68.3
±
0.3%57.8
±
2.7% 油酸,8.8%64.2
±
0.6%53.5
±
6.6% 油酸,5%54.3
±
0.6%68.7
±
7.1% 亚油酸,5%48.2
±
1.6%70.2
±
5.8% 亚麻酸,5%56.2
±
0.6%82.5
±
1.6% 甘油酸三油酸酯,15%28.6
±
1.1%74.3
±
2.2% 甘油酸三油酸酯,5%24.7
±
0.8%81.5
±
4.4% [0141]
由表10可以看出,空白姜黄挥发油-白藜芦醇微乳的包封率~52%,7天药物泄漏率为~76%。当加入15%负油相调节剂后,包封率可提升至80%以上,7天药物泄漏率也可以将低至~45%水平。加入8.8%负油相调节剂后,包封率维持在~65%水平,7天药物泄漏率也可以将低至~55%水平。加入5%负油相调节剂后,包封率回落到50%水平,7天药物泄漏率在在70~80%区间。当加入并不合适的正油相调节剂后,包封率降至~25%,7天药物泄漏率在74~81%区间。
[0142]
可见,根据本发明的方法选择合适的油相调节剂和用量,不仅可以调整合适的微乳尺寸,还能改善所制得的微乳包封率和稳定性。并且,通过本发明所优化制得的姜黄挥发油-白藜芦醇微乳包封率和稳定性与粒子尺寸存在一定的负相关性。
[0143]
同样的增溶改善现象还出现在紫杉醇、长春碱、紫草素、大麻二酚、他克林、大麻萜酚、大麻环酚、cbn大麻酚、二羟基大麻酚、四氢大麻酚、大麻环萜酚、黄芩苷、黄芪甲苷、芍药苷、连翘苷等难溶物研究实例中。
[0144]
实施例10
[0145]
与实施例1区别仅在于,分别选用薏苡仁油一等品、二等品、三等品、随机混合薏苡仁油为油相制备微乳,测试不同油相、比例和油相调节剂制得的乳液的微乳粒径,结果如表11所示:
[0146]
表11亚油酸参与下的不同品级(酸值)薏苡仁油微乳的粒径
[0147]
薏苡仁油品级微乳粒径亚油酸添入比例(wt%)随机混合124.2
±
1.4nm0%随机混合31.6
±
2.4nm15%一等品29.8
±
0.6nm23%二等品32.5
±
0.4nm15%三等品29.8
±
0.6nm6%一等品169.5
±
4.2nm0%三等品109.8
±
2.6nm0%
[0148]
结果如表11所示,随机混合的薏苡仁油微乳粒径在有15%亚油酸和没有掺入的情
况下,粒径分别为~30nm和~120nm。一等品薏苡仁油在有15%亚油酸和没有掺入的情况下,粒径分别为~30nm和~170nm;三等品薏苡仁油在有15%亚油酸和没有掺入的情况下,粒径分别为~30nm和~110nm。同一种类不同品级(酸值)的薏苡仁油,经过式1的启示,可以将相应的微乳粒径控制在30nm水平。
[0149]
作为对比,当各品级薏苡仁油制备的微乳无甘油酸三油酸酯添入时,粒径约为120~170nm范围,当掺入15%甘油酸三油酸酯后,微乳粒径出现不可控的增高,并且澄明度迅速降低,如表12所示:
[0150]
表12甘油酸三油酸酯参与下的不同品级(酸值)薏苡仁油微乳的尺寸调控行为
[0151][0152]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1