一种癌症驱动基因识别方法及系统
1.本发明属于癌症驱动基因识别的技术领域,尤其涉及一种癌症驱动基因识别方法及系统。
背景技术:
2.作为全球第二大常见死因,癌症每年导致超过800万人死亡,预计未来几十年癌症发病率将增加50%以上。体细胞中发生多种基因突变,如单核苷酸变异、结构变异、插入和缺失和拷贝数改变,但大多数突变是随机的。这些对细胞没有功能影响的随机突变被称为乘客突变。还有少数突变增加了细胞相对于其邻居的选择性生长优势,并允许它自行繁殖和侵入其他组织。这些突变可能促进癌症的发生和发展,它们被称为驱动突变。携带驱动突变的基因被认为是驱动基因。一些不含突变但表达异常的基因如果能促进癌症的发展,也可能是驱动基因。在这项发明中,本技术主要关注突变的驱动基因。
3.随着生物数据中信息量的不断增加,越来越多的计算方法可以从基因组数据中准确定位与癌症因果关系相关的基因,这推动了癌症驱动基因的综合鉴定取得了重大进展。基于频率的方法通过研究其突变特征和预设背景突变率来发现癌症驱动基因,因为癌症驱动基因应该在不同样本中频繁变化。然而,背景突变率通常无法正确估计,基于频率的方法往往会忽略突变频率低的致癌驱动基因。基于网络的方法评估了生物网络中的遗传基因,最后通过评估基因在网络中的作用来区分驱动基因,例如drivernet、cbna、netsig等方法,在网络层面识别癌症基因并揭示其分子机制,但结果依赖于所使用的网络方法。
4.随着近年来的发展,机器学习(ml)已成功应用于几个重大的生物医学问题,特别是,机器学习方法因其在生物信息学领域的多项预测任务中的出色表现而受到越来越多的关注。值得注意的是,在许多处理高维数据的机器学习方法中,集成方法通常优于个体分类方法。因此,需要一种基于集成学习的方法来准确的识别癌症驱动基因。
技术实现要素:
5.为能够同时考虑分子特征和网络结构特征,避免单个分类器性能偏差的问题,提高癌症驱动基因的识别准确性,为此,本发明提出了一种癌症驱动基因识别方法及系统,具体方案如下:
6.一种癌症驱动基因识别方法,包括以下步骤:
7.s1、收集正常样本与肿瘤样本的多组学数据,计算出组学特征值和网络结构特征值,所述组学特征值包括差异表达值、差异甲基化值、基因突变频率值、蛋白质-蛋白质相互作用网络数据;
8.s2、将处理后的组学特征值和网络结构特征值进行拼接,构建新的融合特征;另外使用集成学习的方法建立模型,使用序列前向选择策略对模型的第一层分类器进行筛选,第二层分类器采用逻辑回归对第一层分类器的结果赋予权重;
9.s3、将新的融合特征输入到第一层的分类器,获得对应数量的分类器的预测概率;
10.s4、将第一层的所有分类器的预测概率拼接后作为特征输入第二层的逻辑回归分类器进行拟合,最终模型输出表示基因成为驱动基因的概率。
11.具体地说,所述多组学数据包括差异表达值、差异甲基化、基因突变频率值和蛋白质-蛋白质相互作用网络数据;其中基因的表达值、dna甲基化值和基因突变数据来自tcga数据库,蛋白质-蛋白质相互作用网络数据来自consensuspath db数据库。
12.具体地说,在正常样本和肿瘤样本中,只有同时具有可用基因表达值或dna 甲基化值的癌症类型的多组学数据被保留并使用,具体包括8000多个正常样本和16种不同癌症类型的肿瘤样本。
13.具体地说,差异表达值被准确测量为肿瘤表达与配对的正常样本表达之间的log2fold变化,然后在样本中取平均值;所述基因突变频率值为在特定癌症类型的所有样本中观察到的单核苷酸变异和拷贝数变异的平均值,拷贝数变异包括扩增变异和缺失变异。
14.具体地说,计算差异甲基化值由所有肿瘤样本和正常样本的甲基化信号的平均值确定,具体计算利用公式(1)计算得:
[0015][0016]
其中,表示第c种癌症中基因i差异dna甲基化值,和分别是癌变样本和配对的正常样本中的甲基化信号,sc代表一种癌症的样本集。
[0017]
具体地说,网络结构特征值通过蛋白质-蛋白质相互作用网络数据计算获得, mtgcn中使用的蛋白质-蛋白质相互作用网络数据是从consensus path db数据库中收集的,在消除分数小于0.5的交互之后,获得了一个具有13,627个节点和504,378条边的网络,接着利用深度游走算法获取网络结构特征值,深度游走公式为:
[0018][0019]
其中,c
i-1
=v表示当前节点;ci=x表示下一个要到达的节点;其中nv表示节点v的邻居节点,|nv|表示nv的数量。
[0020]
具体地说,构建新的融合特征的具体步骤如下:
[0021]
sa21、将步骤s1中计算得到的差异表达值、差异甲基化值、基因突变频率值后,将每个基因分配到一个n*y维向量,其中n表示基因的数量,y表示组学类型,即差异表达、差异甲基化、基因突变频率;
[0022]
sa22、将n*3维向量连接起来,形成一个n行48列的泛癌矩阵,在连接不同尺度的不同矩阵前,进行最小-最大归一化;
[0023]
sa23、将n行48列的泛癌矩阵和16维的网络结构特征值矩阵直接左右拼接,得到每个基因的融合特征。
[0024]
具体地说,使用序列前向选择sfs策略对第一层分类器进行筛选,第二层分类器采用逻辑回归对第一层分类器的结果赋予权重的具体步骤为:
[0025]
sb21、模型的第一层选择7个分类器,分别为支持向量机、随机森林rf、决策树dt、
多层感知器、极端梯度提升、自适应提升算法和梯度提升gb,使用序列前项选择选择策略,为了实现最好的结果,本技术使用支持向量机、自适应提升算法和多层感知器组合作为第一层的分类器;
[0026]
sb22、第二层分类器来对第一层的分类器性能赋予不同权重,以便更准确的分类,第二层分类器选择逻辑回归lr。
[0027]
实现上述一种癌症驱动基因识别方法的系统,包括
[0028]
收集计算单元,用于收集正常样本与肿瘤样本的多组学数据,计算出组学特征值和网络结构特征值,所述组学特征值包括差异表达值、差异甲基化值、基因突变频率值和蛋白质-蛋白质相互作用网络数据;
[0029]
融合特征构建单元,将处理后的组学特征值矩阵和网络结构特征值矩阵进行左右拼接,构建新的融合特征;
[0030]
集成学习单元,另外建立模型,使用序列前向选择sfs策略对模型的第一层分类器进行筛选,第二层分类器采用逻辑回归对第一层分类器的结果赋予权重;将新的融合特征放入单元中,最终输出表示基因成为驱动基因的概率。
[0031]
一种癌症驱动基因识别方法的实现介质,所述介质存储用于执行上述方法的计算机指令。
[0032]
本发明的有益效果在于:
[0033]
1、本发明更准确地识别癌症驱动基因,可以克服单个分类器性能偏差问题。
[0034]
2、由于典型的驱动基因与非驱动基因在系统和分子层面上的特征存在差异,而本发明同时考虑了分子特征和网络结构特征。
[0035]
3、针对目前存在的识别癌症驱动基因准确率低、单个分类器易出现性能偏差、未同时考虑分子特征和网络结构特征的问题,整合不同类型的特征数据,有效提高预测精度。具体的说是将深度游走算法提取的网络结构特征值与组学特征值相融合,使用集成学习的方法对癌症驱动基因进行预测,有效解决癌症驱动基因识别的准确率低、单个分类器出现性能偏差和未同时考虑分子特征和网络结构特征的问题。
附图说明
[0036]
图1为本发明的总体流程图;
[0037]
图2为组学数据获得流程图;
[0038]
图3为本发明使用的数据的生成过程图;
[0039]
图4和图5分别为十次五折交叉验证下每种方法的受试者工作特征曲线下面积和精确召回曲线下面积;
[0040]
图6为不同方法在两个独立测试集上的性能比较;
[0041]
表1和表2为十次五折交叉验证下每种方法使用网络结构特征前后的受试者工作特征曲线下面积和精确召回曲线下面积。
具体实施方式
[0042]
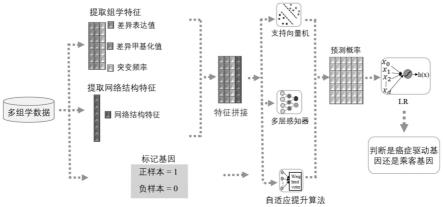
如图1所示,一种癌症驱动基因识别方法,包括以下步骤:
[0043]
s1、收集正常样本与肿瘤样本的多组学数据,计算出组学特征值和网络结构特征
值,所述组学特征值包括差异表达值、差异甲基化值、基因突变频率值;;
[0044]
所述多组学数据包括差异表达值、差异甲基化值、基因突变频率和蛋白质
‑ꢀ
蛋白质相互作用网络数据;其中基因表达、dna甲基化和基因突变数据来自tcga 数据库,蛋白质-蛋白质相互作用网络数据来自consensus path db数据库。
[0045]
在正常样本和肿瘤样本中,只有具有可用基因表达数据值和dna甲基化值的癌症类型的多组学数据被保留并使用,具体包括8000多个正常样本和16种不同癌症类型的肿瘤样本。
[0046]
示例性的,如图2所示,以一种肿瘤为例,在sn个正常样本、肿瘤样本中每个样本均包括gn个基因,对正常样本和肿瘤样本间的基因计算差异表达,得到gn*sn的矩阵,然后针对每行计算每个基因的均值,最后获得gn*1的矩阵,所述值可以为差异表达值和差异甲基化值。基因突变频率值为在特定癌症类型的所有样本中观察到的单核苷酸变异和拷贝数变异的平均值,拷贝数变异包括扩增变异和缺失变异。
[0047]
具体地说,正样本是指已知的癌症遗传基因,包括癌症基因网络(ncg)的权威专家规划的711个已知癌症遗传基因目录,cosmic cgc的超集,以及一组 85个使用digsee从pubmed中提取的高保真癌症遗传基因。负样本是很可能与癌症无关的基因。在该方案中,是通过递归删除与癌症相关的基因来提取负样本,其中主要包括属于ncg、cosmic的遗传基因,kegg数据库中与“癌症通路”相关的遗传基因,omim疾病数据库和mutsigdb中与癌症相关的基因。最后,标准数据集包含796个正样本和2187个负样本。在两个单独的数据集上进行测试,以调查它们的性能是否对特定数据集存在偏差。这两个独立的测试集来自mtgcn。收集了来自oncokb的1064个癌基因和来自ongene的803个癌基因。筛选出这些基因和泛癌数据集的共同基因,然后从这些共同基因中去除用于训练的标记样本。最后,oncokb数据库包含320个癌症基因,ongene数据库包含388个癌症基因。
[0048]
以下差异表达值、差异甲基化值、基因突变频率值、蛋白质-蛋白质相互作用网络数据计算步骤分别如下:
[0049]
(1)差异表达值
[0050]
差异表达值被准确测量为肿瘤表达与配对的正常样本表达之间的log2fold 变化,然后在样本中取平均值。
[0051]
(2)差异甲基化值
[0052]
计算差异甲基化值由所有肿瘤样本和正常样本的甲基化信号的平均值确定,具体计算利用公式(1)计算得:
[0053][0054]
其中,表示第c种癌症中基因i差异dna甲基化值,和分别是癌变样本和配对的正常样本中的甲基化信号,sc代表一种癌症的样本集。
[0055]
(3)基因突变频率值
[0056]
基因突变频率值代表在特定癌症类型的所有样本中观察到的单核苷酸变异和拷贝数变异的平均值,具体而言,拷贝数变异包括扩增变异和缺失变异。
[0057]
(4)网络结构特征值
[0058]
网络结构特征值通过蛋白质-蛋白质相互作用网络数据计算获得,mtgcn 中使用
的蛋白质-蛋白质相互作用网络数据是从consensus path db数据库中收集的,在消除分数小于0.5的交互之后,获得了一个具有13,627个节点和504,378 条边的网络,接着利用深度游走算法获取网络结构特征值,深度游走公式为:
[0059][0060]
其中,c
i-1
=v表示当前节点,ci=x表示下一个要到达的节点。其中nv表示节点v的邻居节点,|nv|表示nv的数量。换句话说,每个邻居节点都具有相同的被选中概率。深度游走提取的特征维数设置为16。
[0061]
s2、将处理后的组学特征值和网络结构特征值进行拼接,构建新的融合特征;另外使用集成学习的方法,建立模型,使用序列前向选择sfs策略对模型的第一层分类器进行筛选,第二层分类器采用逻辑回归对第一层分类器的结果赋予权重;
[0062]
构建新的融合特征的具体步骤如下:
[0063]
sa21、将步骤s1中计算得到的差异表达值、差异甲基化值、基因突变频率值后,将每个基因非配到一个n*y维向量,其中n表示癌症类型的数量,y表示组学类型,即差异表达、差异甲基化、基因突变频率,在该方案中为16*3维向量;
[0064]
sa22、将n*3维向量连接起来,形成一个n行48列的泛癌矩阵,需要注意的是,在连接不同尺度的不同矩阵前,进行最小-最大归一化。
[0065]
sa23、将n行48列的泛癌矩阵和16维的网络结构特征值矩阵直接左右拼接,得到每个基因的融合特征。
[0066]
使用序列前向选择sfs策略对第一层分类器进行筛选,第二层分类器采用逻辑回归对第一层分类器的结果赋予权重的具体步骤为:
[0067]
sb21、模型的第一层选择7个分类器,分别为支持向量机、随机森林、决策树dt、多层感知器多层感知器、极端梯度提升、自适应提升算法和梯度提升,使用序列前项选择选择策略,为了实现最好的结果,本技术使用支持向量机、自适应提升算法和多层感知器组合作为第一层的分类器;
[0068]
sb22、第二层分类器来对第一层的分类器性能赋予不同权重,以便更准确的分类,第二层分类器选择逻辑回归lr。
[0069]
如图3所示,最后将每个基因的48维生物学特征和16维结构特征结合起来,得到每个基因的融合特征。
[0070]
s3、将新的融合特征输入到第一层的分类器,获得对应数量的分类器的预测概率;在该方案中得到3个分类器的预测概率。
[0071]
s4、将第一层的所有分类器的预测概率拼接后作为特征输入第二层的逻辑回归分类器进行拟合,最终模型输出表示基因成为驱动基因的概率。
[0072]
为评估模型的性能,将结果与单个分类器和其他方法进行比较,评价指标为: (i)受试者工作特征曲线下面积(auroc),和(ii)精确召回曲线下面积 (auprc)。auc为受试者工作特征曲线下面积,这个面积的数值介于0到1 之间,能够直观的评价出分类器的好坏,auc的值越大,分类器效果越好,其计算公式如下:
[0073][0074]
其中,代表第i个样本的序号(概率得分从小到大排,排在第rank个位置),m、n分别是正样本和负样本的个数,指将正样本的序号相加。对所有标记的数据运行十次五折交叉验证,计算平均值作为模型结果。
[0075]
图4、5分别展示了本技术的模型与单个分类器、其他方法的性能比较。图 4结果表明,本技术的模型比单一学习算法具有更高的预测精度,验证了本技术提出的集成方法的有效性。图5展示了所提出的模型略优于mtgcn,但它很容易实现,因为它仅基于经典分类器,不需要过多的调整或特征选择,表明了模型的有效性和优越性。
[0076]
为了更好地检测网络结构特征是否有利于提高预测结果,本技术在仅使用分子特征和添加网络结构特征后,依次将本技术的模型与单一分类器、其他方法进行比较。从表1、2可以看出,在所有情况下,使用融合特征都比单独使用分子特征表现得更好,证明了网络结构特征的有效性。表明分子特征和网络结构特征相结合可以提高模型的预测准确性。
[0077]
在两个单独的数据集上测试本技术的模型和其他方法,使用所有泛癌阳性和阴性样本来训练本技术的模型和其他方法,将来自两个独立测试集的基因放入已经训练好的模型中进行预测。图6显示了来自oncokb数据库(x轴)和 ongene数据库(y轴)的两个不同独立驱动基因集的精确召回曲线下面积比较。所有方法的精确召回曲线下面积都非常小,因为真阳性基因的总数很低。可以注意到,提出的模型在两个独立的测试集上都优于其他方法。
[0078]
实现上述的一种癌症驱动基因识别方法的系统,包括
[0079]
收集计算单元,用于收集正常样本与肿瘤样本的多组学数据,计算出组学特征值和网络结构特征值,所述组学特征值包括差异表达值、差异甲基化值、基因突变频率值;
[0080]
融合特征构建单元,将处理后的组学特征值和网络结构特征值进行拼接,构建新的融合特征;
[0081]
集成学习单元,另外建立模型,使用序列前向选择sfs策略对模型的第一层分类器进行筛选,第二层分类器采用逻辑回归对第一层分类器的结果赋予权重;将新的融合特征放入单元中,最终输出表示基因成为驱动基因的概率。
[0082]
本技术实施例提供了一种癌症驱动基因识别方法的实现介质,所述介质存储用于执行上述开发方法的计算机指令。
[0083]
本技术中的各个实施例均采用递进的方式描述,各个实施例之间相同相似的部分互相参见即可,每个实施例重点说明的都是与其他实施例的不同之处。尤其,对于系统和介质实施例而言,由于其基本相似于方法实施例,所以描述的比较简单,相关之处参见方法实施例的部分说明即可。
[0084]
本技术实施例提供的系统和介质与方法是一一对应的,因此,系统和介质也具有与其对应的方法类似的有益技术效果,由于上面已经对方法的有益技术效果进行了详细说明,因此,这里不再赘述系统和介质的有益技术效果。
[0085]
本领域内的技术人员应明白,本技术的实施例可提供为方法、系统、或计算机程序产品。因此,本技术可采用完全硬件实施例、完全软件实施例、或结合软件和硬件方面的实施例的形式。而且,本技术可采用在一个或多个其中包含有计算机可用程序代码的计算机
可用存储介质(包括但不限于磁盘存储器、cd
ꢀ‑
rom、光学存储器等)上实施的计算机程序产品的形式。
[0086]
本技术是参照根据本技术实施例的方法、设备(系统)、和计算机程序产品的流程图和/或方框图来描述的。应理解可由计算机程序指令实现流程图和/或方框图中的每一流程和/或方框、以及流程图和/或方框图中的流程和/或方框的结合。可提供这些计算机程序指令到通用计算机、专用计算机、嵌入式处理机或其他可编程数据处理设备的处理器以产生一个机器,使得通过计算机或其他可编程数据处理设备的处理器执行的指令产生用于实现在流程图一个流程或多个流程和/或方框图一个方框或多个方框中指定的功能的装置。
[0087]
这些计算机程序指令也可存储在能引导计算机或其他可编程数据处理设备以特定方式工作的计算机可读存储器中,使得存储在该计算机可读存储器中的指令产生包括指令装置的制造品,该指令装置实现在流程图一个流程或多个流程和/或方框图一个方框或多个方框中指定的功能。
[0088]
这些计算机程序指令也可装载到计算机或其他可编程数据处理设备上,使得在计算机或其他可编程设备上执行一系列操作步骤以产生计算机实现的处理,从而在计算机或其他可编程设备上执行的指令提供用于实现在流程图一个流程或多个流程和/或方框图一个方框或多个方框中指定的功能的步骤。
[0089]
在一个典型的配置中,计算设备包括一个或多个处理器(cpu)、输入/输出接口、网络接口和内存。内存可能包括计算机可读介质中的非永久性存储器,随机存取存储器(ram)和/或非易失性内存等形式,如只读存储器(rom)或闪存 (flash ram)。内存是计算机可读介质的示例。
[0090]
计算机可读介质包括永久性和非永久性、可移动和非可移动媒体可以由任何方法或技术来实现信息存储。信息可以是计算机可读指令、数据结构、程序的模块或其他数据。
[0091]
计算机的存储介质的例子包括,但不限于相变内存(pram)、静态随机存取存储器(sram)、动态随机存取存储器(dram)、其他类型的随机存取存储器(ram)、只读存储器(rom)、电可擦除可编程只读存储器(eeprom)、快闪记忆体或其他内存技术、只读光盘只读存储器(cd-rom)、数字多功能光盘(dvd)或其他光学存储、磁盒式磁带,磁带磁磁盘存储或其他磁性存储设备或任何其他非传输介质,可用于存储可以被计算设备访问的信息。按照本文中的界定,计算机可读介质不包括暂存电脑可读媒体(transitory media),如调制的数据信号和载波。
[0092]
本技术主要用于支持局域网或广域网环境下的大规模系统仿真,满足仿真应用规模、结构日益扩大和复杂的需求,是具有分布、异构、协同、互操作、重用等性能的分布式仿真运行支撑环境构建技术。
[0093]
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1