不良事件数据采集管理系统的制作方法
1.本发明具体涉及一种不良事件数据采集管理系统。
背景技术:
2.随着科技的发展,越来越多的疾病被新药治愈,而新药研发是一个漫长的过程,因其特殊性,新药自研发到批准上市的过程中需要进行大量的测试和多次的临床实验,以最大程度降低新药对人体的损伤,而药品测试实验过程信息采集和分析不良事件尤为重要,不良事件,指受试者接受试验用药品后出现的所有不良医学事件,可以表现为症状体征、疾病或者实验室检查异常,不良事件的存在不一定与试验用药品有因果关系,因此需要记录和分析大量的不良事件,对药品进行改良避免不良事件的发生,提高药物的安全性和有效性;因临床实验周期长,中间环节多所以会产生大量的数据,需要根据采集到的信息进行不良事件的判定,而针对不良事件数据的采集、采集资料管理和实验过程管理难度颇大,因此需要一个操作系统来解决这些问题。
3.在中国专利:202011093592 .8,名称为:一种临床科研数据采集管理系统中记载有包括:s1.完成方案搭建,在进行数据采集分析之前建立好数据分析模型,根据项目方案,完成项目创建所需的基本信息的填写、访视周期的配置,并建立访视计划,最后完成crf配置;s2 .新建受试者,对受试者的基本信息进行收录以及对受试者后期的随访进行数据采集;s3 .自动生成随访计划,对受试者以通讯或其他的方式,进行定期了解病情变化和指导患者康复;s4.进行数据采集,将受试者的信息进行数据管理和数据溯源;s5.进行数据查询、分析、导出并对导出数据进行报表管理。
4.如专利202011093592 .8中图1所示,该方案关于受试者信息的采集还是通过定期对受试者进行随访,然后再由随访者采集填写受试者用药后信息从而进行不良事件的判定,这种方式在两个随访日期的中间存在空白时间段,不能及时获取受试者用药后各种身体状况,不利于测试项目的进行,也存在对受试者一些信息采集遗漏的可能,导致一些不良事件未被监测到,另外该方案不具备监督管理功能,对测试过程违规和超出测试计划的非正常情况不能记录和完全掌控,导致记录测试数据和测试结果的真实度和准确度存在不确定性。
技术实现要素:
5.本发明所要解决的技术问题是,技术背景中提及的现有系统不良事件信息采集录入不及时,和对测试过程中无监督管理容易导致记录测试数据和测试结果的真实度和准确度存在不确定性的问题。
6.针对上述技术问题,提出一种不良事件数据采集管理系统;通过以下技术方案实现的:一种不良事件数据采集管理系统,包括储存模块,包括系统指令数据,受试人员信息,测试药物信息,系统运行配置文件和数据分析模型;录入模块,包括对受试人员的档案建立和测试各个过程中受试人员健康状况填写
以及自动对受试人员编写身份识别码;信息采集模块,包括建立信息采集标准和信息采集项,建立信息采集计划流程,按照计划给受试人员和信息采集人员推送当日任务提醒,由受试人员和信息采集人员分别填写需采集信息,并向受试人员和信息采集人员发送未完成任务提醒,受试人员未在规定时间内完成信息填写的本人不可再填写,由信息采集人员根据实际情况完成补录,并填写违规原因;处理分析模块,包括对受试人员和信息采集人员每日任务提交的信息进行分析处理,生成数据报表,对不良事件进行判定和评级,并向研究医生发送不良事件提醒;仓储模块,包括对所有测试药物的自动编写药物码和记录各种药物的库存使用情况,并且每盒出库的药物均要对应到相应的受试人员,做到每盒出库药物和受试人员对应的唯一性,从而做到从药品入库到受试人员使用全流程的管控;监查模块,根据受试人员身份识别码查询该受试人员使用药物记录和用药前后身体健康指标查询,或根据出库后的任意一种药物的药物码查询药物对应受试人员及出入库记录,对使用药物进行溯源,并且监查模块链接监督管理机构,记录监管整个测试过程、测试数据和测试结果保障实验数据的真实性。
7.本发明,按照设定好的计划,利用信息采集模块每日向受试人员和信息采集人员推送当日任务提醒,提醒受试人员填写当日日记卡和需采集的信息,并且由信息采集人员进行确认和提交,保证了数据录入的及时性,另外监查模块链接监督管理机构,对整个测试过程、测试数据和测试结果进行记录监管,并且记录内容的不可更改性,保证了测试数据和测试结果的真实性。
8.对本发明技术方案的优选,所述不良事件数据采集管理系统,支持登录人员权限设置,给予不同登录人员不同查看权限,低级权限平台操作界面不显示超过本权限范围内的操作键,不同登录人员不同权限的设置,使得每个人的操作界面只有自己负责的模块,使得操作更简捷高效,节约时间。
9.对本发明技术方案的优选,信息采集模块支持多人同时登录在线操作,支持多人同时登录在线操作的设置便于受试人员和信息采集人员填写需要采集的信息,便于操作的同时也加快了不良事件数据的采集,加快了实验进程。
10.对本发明技术方案的优选,受试人员和信息采集人员填写的采集信息系统每日自动保存并自动传入处理分析模块进行分析处理,采集信息自动上传分析的设置使得不良事件数据采集管理系统的人机信息交互更友好,减轻了数据分析的工作量,也加快了对采集数据的分析速度。
11.对本发明技术方案的优选,所述录入模块支持信息加密,能对录入的受试人员的姓名电话及家庭住址进行加密隐藏,录入后隐藏患者身份信息,仅以受试人员身份识别码作为显示和区分,并且对加密隐藏信息的读取设置读取权限,只有系统指定级别的权限才能查看此类信息,受试人员信息加密的设置保护了受试人员的个人隐私,同时指定级别权限查看加密信息的设置使得,当遇到紧急情况时,通过高权限可查到受试人员信息,从而紧急联系受试人员。
12.对本发明技术方案的优选,录入模块自动编写的身份识别码和仓储模块自动编写的药物码具有唯一性,不重复,身份识别码和药物码唯一性的设置使得药物和受试人员之
间的关联具有唯一性,确保每份药剂的使用者的唯一性,便于每份药剂的使用过程追溯,同时也使得实验数据和过程记录更精确,提升了实验的严谨度。
13.对本发明技术方案的优选,监查模块记录测试过程违规和超出测试计划的非正常情况,记录测试数据和测试结果,并且监查模块记录的内容均不可更改,监查模块的设置便于监管机构监督实验过程中操作规范的情况,同时监查模块记录的测试数据和测试结果不可修改的设置,能有效避免数据造假,保证了实验数据和结果的准确性。
14.本发明与现有技术相比具有的有益效果是:本发明的技术方案,按照设定好的计划,利用信息采集模块每日向受试人员和信息采集人员推送当日任务提醒,提醒受试人员填写当日日记卡和需采集的信息,并且由信息采集人员进行确认和提交,保证了数据录入的及时性,另外监查模块链接监督管理机构,对整个测试过程、测试数据和测试结果进行记录监管,并且记录内容的不可更改性,保证了测试数据和测试结果的真实性。
附图说明
15.图1为不良事件数据采集管理系统的流程示意图。
16.图2为信息采集模块的流程示意图。
17.图3为仓储模块的流程示意图。
18.图4为处理分析模块的流程示意图。
具体实施方式
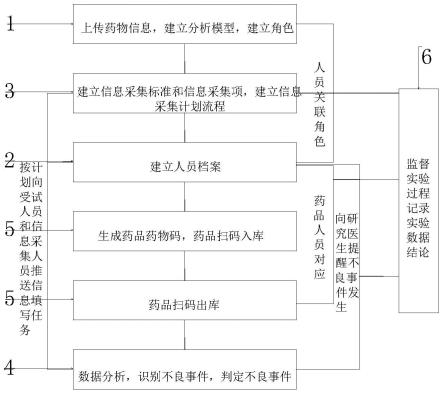
19.下面将结合本发明实施例中的附图1-4,对本发明实施例中的技术方案进行详细的描述。
20.实施例1本文使用的“受试人员”是指参加实验的健康志愿者或患者。
21.本文使用的“信息采集人员”是指实验过程中除受试人员外,用于采集受试人员信息的其他人员,可以是受试人员检查医生。
22.本文使用的“研究医生”是指执行临床实验的医生。
23.本文使用的“监督管理人员”是指监督实验过程规范的人员,可以是药品监督管理局监管新药测试人员也可是官方认可的第三方机构。
24.本文使用的“系统工程师”是指对系统足够了解,维护系统的工程师。
25.如图1、2、3和4所示,一种不良事件数据采集管理系统,包括储存模块1、录入模块2、信息采集模块3、处理分析模块4、仓储模块5和监查模块6。
26.储存模块1主要是用于储存系统的指令数据,配置文件,分析模型,人员和药物信息等一系列需要存储的文件,另外储存模块1内单独分区出一块用于备份系统数据,且备份工作会在每天凌晨自动备份,不影响白天使用的同时也能防止数据丢失,当发生一些不可控事件导致系统数据丢失情况时,可由系统工程师从后台用备份资料恢复系统数据,使得系统数据存储更安全。
27.录入模块2主要用于系统配置文件上传和人员档案建立,此处人员包括受试人员,信息采集人员,研究医生和监管机构监督管理人员,另外为了保护参加测试人员的隐私信
息,录入模块2会自动对建档人员的个人信息进行加密,并且自动生成一个身份识别码,利用身份识别码登录系统,并且可针对每个身份识别码单独设置系统登录密码,或设置统一登录密码,而后由个人自行修改,为了防止密码忘记,修改后的密码在系统后台可重置。
28.为了增强录入模块2的录入速度减小工作量,录入模块2可通过表格上传录入信息,上传信息所用的表格模板由系统生成,表格内包含系统识别的格式,只需要按照表格要求填写上传信息即可。
29.为了便于管理和后续人员权限的建立,在录入模块2建立人员档案前,先建立人员角色,把后续建立档案的每个人员都归纳到相对应的角色内,之后对角色分配权限建立的角色有多个分别为受试人员、信息采集人员、研究医生、监督管理人员和系统工程师,角色可根据实际需要自行建立,在填写人员档案建立的表格时,把每个人员对应的角色要填上后再通过录入模块2上传表格,建立人员档案。
30.为了简化不同角色对系统的操作步骤,提升数据采集效率和降低工作难度,系统对不同角色赋予不同的权限,不同角色按照职责分配权限,低权限角色操作界面不显示高于自身权限的所有资料和信息,不同角色按照权限各司其职,简化工作流程提高办事效率。
31.仓储模块5主要用于对药物的管理,所有的测试药物要先录入仓储模块5进行备案和入库,入库后的测试药物方可分发给受试人员,并且系统会为入库后的每份药物自动编订药物码,入库后的药物会连接该药物所对应的计划,当药物出库使用后,药物对应的计划会启动,系统会把计划对应的每日任务推送给对应的受试人员。
32.关于测试药物的入库和分发的管理,测试药物制好后统一先入库,先按照数量由系统自动生成药物码,然后把药物码分别贴在每份药剂盒上,贴好后统一扫描药物码对药物扫描入库,由于药物种类较多,可在仓储模块5中对仓库进行分区,不同药剂存放在不同分区,方便查找取用,当需要药剂出库分发给受试人员时,首先在出库界面扫描要出库的药物识别码,然后再扫描受试人员的身份识别码,然后系统会按照药物对应的计划向该受试人员推送每日任务,由受试人员按照时间登陆系统,填写需要采集的数据信息,因此仓储模块5每份出库的药剂都对应着唯一的一个受试人员,所以能对药剂从入库到受试人员使用过程的全流程管控,保证了药剂和受试人员连接关系,保证了后续信息数据采集的精准度。
33.信息采集模块3内建立有信息采集标准和信息采集项,信息采集标准和需要采集的信息采集项建立在信息采集前,然后根据实际情况建立信息采集计划,由信息采集模块3按照计划每日向受试人员和信息采集人员推送信息采集任务,由受试人员和信息采集人员按照任务填写需要采集的信息,完成数据的信息采集,填写好的信息数据经保存后会自动传入处理分析模块4,由处理分析模块4进行处理分析,判定是否存在不良事件,并且对不良事件进行评级,最后把不良事件结果和提醒发至对应的研究医生。
34.关于数据信息的采集,在药剂出库时就已经绑定对应的受试人员,受试人员在规定时间内使用药物,药剂出库时会启动该药剂对应的信息数据采集计划,并且把该药剂阶段的信息数据采集计划绑定使用该药剂的受试人员,在计划开始后按照计划向受试人员发送每日任务,由受试人员按照自己的身份识别码登录系统,按照自身用药后情况填写实验需要收集的数据,数据填写完成后受试人员提交任务,提交后的任务坏会先发送到信息采集人员,由信息采集人员对受试人员填写的数据进行查验,和确认,以起到对突发事件的紧急处理,另外若受试人员未在规定时间内填写和提交信息数据采集任务的,系统会通知受
试人员和信息采集人员,督促监督完成任务,并且为了保证实验的顺利进行,受试人员未在规定时间内完成信息填写的本人不可再填写,由信息采集人员根据实际情况完成补录,并填写违规原因,系统会记录该违规事件,按照实验规范对事件进行处理。
35.另外为了更方便的收集受试人员的身体指标,在受试人员定期的检查中,系统会向受试人员推送检查提醒,提醒受试人员在规定时间内向指定医院进行身体指标检测,检测结果由负责医生通过拍照或扫描录入系统,由分析模块4进行分析处理。
36.处理分析模块4主要用于对受试人员和信息采集人员提交和收集到的信息进行分析处理,由分析模块4判定受试人员是否存在不良事件,和对发生的不良事件进行评级和提醒,实验结束后,处理分析模块4可根据数据情况生成数据报表,方便查看实验结果,了解药剂特性。
37.关于不良事件的判定和评级,不良事件的判定由处理分析模块4完成,模块根据受试人员提交的信息数据和正常人以及受试人员用药前的信息数据进行对比,和差异值的比较,进而判断受试人员是否发生不良事件,进而再根据标准对不良事件进行评级,关于不良事件的评级可按照标准分为1级-5级,例如依据nci ctcae 5.0标准判断不良事件严重程度,内容可见下方表格。
不良事件分级严重程度描述1级:轻度,无临床症状或有轻微临床症状;仅为临床或诊断所见;无需治疗。2级:中度,需要较小的、局部的或非侵入性治疗;与年龄相当的工具性日常生活活动受限,工具性日常生活活动指做饭、购买衣物、使用电话、理财等。3级:重度或者具重要医学意义但不会立即危及生命;导致住院或者延长住院时间;致残;自理性日常生活受限。自理性日常生活指:洗澡、穿脱衣、吃饭、盥洗、服药等,并未卧床不起。4级:危及生命,需要紧急治疗。5级:与不良事件相关的死亡。
38.当不良事件发生后,处理分析模块4把不良事件的情况和判定结果发送至研究医生,并且记录保存此次不良事件发送至检查模块6进行存档,然后系统把受试人员归类至发生不良事件组类,暂停原计划任务推送,由研究医生查看受试人员的姓名联系方式等被加密隐藏资料,与受试人员联系,对受试人员进行治疗,保障受试人员的安全,并且对此次不良事件进行分析处理,最后决定该受试人员是否继续参与实验,若参与则选定后续推送计划节点,若停止实验则系统停止对该受试人员的后续任务,对该受试人员的数据资料进行封存,做后期对比。
39.监查模块6主要是用于对整个实验过程进行监督和管理,以及对受试人员使用药物记录和用药前后身体健康指标查询,和出入库的药物流向,保证新药实验过程数据的真实性,保证最后新药测试结果的真实性,防止数据造假。
40.关于实验过程的监督,监查模块6对实验全流程监督,记录实验过程中受试人员的数据信息,记录实验过程中存在的违规事件和不良事件,并且对每件不良事件全程记录监控,支持对每份药剂溯源,在实验结束后生成数据报表,确定实验的规范性,准确性和合理性,并且为保障测试的真实性,监查模块6内记录和生成的数据均不可更改。
41.本实施例的使用过程:在一个新药测试实验开始前,首先通过录入模块2向储存模块1内上传测试药物信息和数据分析模型,以及建立相应的实验过程中的参与角色,然后建立信息采集标准和信息采集项,建立信息采集计划流程,而后再建立人员档案,和测试药品
的编码入库,而后再分发实验药物至受试人员,并且实验药物和受试人员一一对应,而后再根据药物和受试人员的对应关系,向受试人员按计划推送当日的任务,采集用药后信息数据,而后再由处理分析模块4对采集到的数据进行分析处理判定不良事件的是否发生和发生不良事件的评级,并将不良事件通知到研究医生,由研究医生进行处理记录系统,并且再决定该受试人员是否继续试验,最后由处理分析模块4分析处理生成报表,为了便于对受试人员使用的药物溯源,和保证试验数据和试验结果的真实性,利用监查模块6对实验全流程监督,并且实验结束是出具监督报告,记录实验过程中是否存在不规范和违规操作。
42.以上实施例仅为说明本发明的技术思想,不能以此限定本发明的保护范围,凡是按照本发明提出的技术思想,在技术方案基础上所做的任何改动,均落入本发明保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1