一种引导骨组织再生膜材料、其制备方法及应用
1.本发明涉及生物材料技术领域,具体而言,涉及一种引导骨组织再生膜材料、其制备方法及应用。
背景技术:
2.临床常见患者因牙齿龋坏、根尖周炎、口腔颌面部外伤、肿瘤等各种原因导致患区牙槽骨发生不可逆的骨吸收,牙槽骨吸收后造成的牙槽嵴高度不足,很难满足临床种植修复的要求,使得后期修复变得十分困难。一直以来,自体骨被认为是修复颌面部骨缺损的黄金法则,但由于其来源有限,而且会引起供区不可逆的骨结构破坏的问题;异种或异体骨供体的来源也十分有限,并且可能会引起受者发生免疫排斥反应,因此如何能够既减少患者痛苦并增加骨量,又能保证成功率和效果是现种植医生迫切要解决的问题。
3.引导骨组织再生(guided bone regeneration)技术,又称gbr技术,可以有效的解决这一问题,其因创伤小、骨增量效果好、适应症广泛、操作方便而被广泛应用于口腔种植术中。gbr技术是利用屏障膜阻止成纤维细胞进入骨缺损区,为成骨细胞提供充足的成骨空间,最终达到组织再生定向修复的目的。hurley等在1959年进行实验性脊柱融合治疗时首次提出gbr屏障膜的概念,然而未能将gbr屏障膜技术应用于临床。20世纪80年代初期,karring和nyman在进行了大量针对屏障膜的实验和临床研究后,才认为屏障膜可以用于修复骨缺损,1989年dahlin等将骨组织再生(guided bone regeneration,gbr)技术首次引入到口腔种植手术中,该技术利用gbr膜将骨缺损区域与外界隔绝起来,形成封闭的成骨区,gbr膜可以有效的防止成纤维细胞进入骨缺损区,而让成骨细胞在骨缺损区内可以得到充分的增殖、分化及矿化。
4.gbr技术的改良可通过对gbr膜材料进行改良来实现。常用的gbr膜可分为两大类:不可吸收膜和可吸收膜。其中不可吸收膜虽然可以提供高体积稳定性,然而在植入牙种植体之前或形成新骨后,通常需要进行第二次手术取出,这意味着它会增加感染、疼痛和愈合期的风险,另外材料的刚性可能导致愈合过程中伤口开裂膜暴露,适应性差,可能导致感染和周围纤维组织内生等临床问题。现临床常用的可吸收膜主要有胶原膜、聚乳酸膜和其他高分子合成膜等,其可降解吸收特性则弥补了以上缺陷,并显示出良好的生物相容性和生物吸附性以及射线可穿透性。但是,可吸收膜存在机械强度和刚度不足,其降解速率不定,不能提供适当的体积稳定性,如果膜吸收太快,缺陷区域将没有足够的支撑,尤其是在大面积骨缺损重建中,骨生长的空间将减少。
5.因此,亟需研发一种新型的gbr膜,并且应具有以下的特点:
6.(1)较高机械强度以及空间维持性强,可以有效防止gbr膜破裂,在一定程度上具备可塑性,可以根据不同骨缺损形态塑形,为骨再生提供必要的空间;
7.(2)gbr膜应具备良好的生物相容性;
8.(3)膜的降解时间应当与骨再生时间相适应,如果降解速度过快,就不能长期有效维持骨再生所需的空间,但是降解速度过慢,则会发生不必要的排异或炎症反应;
9.(4)具备细胞屏障性,阻止成纤维细胞迁移到骨缺损区,同时允许成骨所需的营养物质进入。
10.鉴于此,特提出本发明。
技术实现要素:
11.本发明的目的在于提供一种引导骨组织再生膜材料及其制备方法,旨在使制备得到的引导骨组织再生膜材料具有较高空间维持性和可塑形性,同时降解时间和骨再生时间能够相适应。
12.本发明的另一目的在于提供上述引导骨组织再生膜材料在制备口腔骨缺损区修复材料中的应用。
13.本发明是这样实现的:
14.第一方面,本发明提供一种引导骨组织再生膜材料,其包括plga纤维膜,在plga纤维膜孔隙和表面均附着有水凝胶光固化材料;
15.其中,水凝胶光固化材料的原料包括:聚乙二醇二丙烯酸酯、甲基丙烯酸酰化明胶和粘土。
16.在可选的实施方式中,聚乙二醇二丙烯酸酯、甲基丙烯酸酰化明胶和粘土的质量比为10:0.5-1.5:1.0-2.0;
17.优选地,粘土为纳米粘土,粒径为20-100nm;
18.优选地,水凝胶光固化材料在引导骨组织再生膜材料中的质量占比为20-50%;
19.优选地,plga纤维膜是通过静电纺丝的方式制备,其厚度为0.5-2mm。
20.第二方面,本发明提供一种前述实施方式中任一项引导骨组织再生膜材料的制备方法,将plga纤维膜浸泡在由聚乙二醇二丙烯酸酯、甲基丙烯酸酰化明胶和粘土制备得到的光固化水凝胶中,然后将附着有光固化水凝胶的plga纤维膜进行紫外照射。
21.在可选的实施方式中,plga纤维膜的制备过程包括:制备plga静电纺丝溶液,采用静电纺丝的方法制备plga纤维膜。
22.在可选的实施方式中,将plga颗粒和有机溶剂混合均匀后进行脱气处理得到plga静电纺丝溶液,将plga静电纺丝溶液注射至附着在接收滚筒上的接收膜材上形成静电纺丝纤维膜;
23.优选地,控制纺丝电压为10-20kv,纺丝距离为10-20cm,滚筒转速为700-900转/min,注射速率为3-5ml/h;纺丝期间温度控制在22-25℃,湿度为40%-60%;
24.优选地,将制备完成的plga纤维膜置于通风橱中30-60h;
25.优选地,接收膜材选自锡箔、无纺布中的至少一种。
26.在可选的实施方式中,plga静电纺丝溶液的浓度为0.1-0.3g/ml;
27.优选地,plga颗粒制备过程中控制聚乳酸和羟基乙酸的质量比为2-4:1;
28.优选地,有机溶剂选自1,1,1,3,3,3-六氟异丙醇、n,n-二甲基甲酰胺和四氢呋喃中的至少一种。
29.在可选的实施方式中,光固化水凝胶的制备过程包括:将聚乙二醇二丙烯酸酯和水混合得到pegda水凝胶,将pegda水凝胶与甲基丙烯酸酰化明胶颗粒混合得到pegda-gelma混合水凝胶,将pegda-gelma混合水凝胶与粘土混合后再与光引发剂混合;
30.优选地,将pegda水凝胶与甲基丙烯酸酰化明胶颗粒混合,在40-80℃的条件下搅拌10-30min,搅拌转速为800-1500转/min。
31.在可选的实施方式中,聚乙二醇二丙烯酸酯、甲基丙烯酸酰化明胶颗粒和粘土的质量比为10:0.5-1.5:1.0-2.0;每毫升水对应聚乙二醇二丙烯酸酯的用量为0.2-0.4g;每毫升水对应光引发剂的用量为0.0005-0.002g。
32.在可选的实施方式中,将plga纤维膜浸泡在光固化水凝胶中,待其完全浸透后从光固化水凝胶中取出,采用紫外灯在样品每侧均照射20-60s进行紫外光交联;
33.优选地,紫外光的波长为300-400nm,强度为1200-2000w/cm2;
34.优选地,在浸泡之前,将plga纤维膜剪成所需形状。
35.第三方面,本发明提供前述实施方式中任一项引导骨组织再生膜材料或前述实施方式中任一项制备方法制备得到的引导骨组织再生膜材料在制备口腔骨缺损区修复材料中的应用。
36.本发明具有以下有益效果:通过在plga纤维膜上附着由聚乙二醇二丙烯酸酯、甲基丙烯酸酰化明胶和粘土光固化形成的水凝胶光固化材料,制备得到的复合材料具备较高的空间维持性,具有较慢的降解性能,有利于骨缺损区成骨细胞的粘附、增殖和分化,促进骨缺损区的骨再,为缺损区的成骨提供足够的空间和时间条件,以满足种植手术的要求。
37.此外,本发明较佳的实施例中,利用静电纺丝和光固化水凝胶结合,实现了可塑形性,临床中根据不同骨缺损而行个性化的gbr术。
附图说明
38.为了更清楚地说明本发明实施例的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,应当理解,以下附图仅示出了本发明的某些实施例,因此不应被看作是对范围的限定,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他相关的附图。
39.图1为本发明实施例提供制备方法的工艺流程图;
40.图2为实施例和对比例制备得到产品的电镜图和eds图;图2中a表示plga film,b表示plga film-p、c表示plga film-pg、d表示plga film-pgn;
41.图3为实施例和对比例得到产品的热重测试结果图;
42.图4为实施例和对比例得到产品的接触角测试结果图;
43.图5为实施例和对比例得到产品的溶胀试验测试结果图;
44.图6为实施例和对比例得到产品体外降解测试时降解量随时间的变化图(pbs);
45.图7为实施例和对比例得到产品体外降解测试时降解量随时间的变化图(溶菌酶);
46.图8为实施例和对比例得到产品体外降解测试时ph值的变化图;
47.图9为实施例和对比例得到产品力学性能测试时的拉伸应力-应变图;
48.图10为实施例和对比例得到产品力学性能测试时的拉伸力测试结果图;
49.图11为实施例和对比例得到产品力学性能测试时的拉伸模量测试结果图;
50.图12为实施例得到产品空间维持性和可塑形测试过程图;图12中a和b表示光照前后的实物图;c和d表示承受50g外力的测试实验图;
51.图13为实施例和对比例得到产品对bmsc细胞cck-8试验的结果图;
52.图14为实施例和对比例得到产品对l929细胞cck-8试验的结果图;
53.图15为实施例和对比例得到产品对bmsc细胞粘附结果图(1天);
54.图16为实施例和对比例得到产品对bmsc细胞粘附结果图(3天);
55.图17为实施例和对比例得到产品体外成骨试验中对alp试验结果的光镜图;
56.图18为实施例和对比例得到产品体外成骨试验中对alp试验定量结果图;
57.图19为实施例和对比例得到产品体外成骨试验中对茜素红试验结果的光镜图;
58.图20为实施例和对比例得到产品体外成骨试验中对茜素红试验定量结果图;
59.图21为实施例和对比例得到产品细胞屏障作用的测试结果图;
60.图22为实施例和对比例得到产品体内成骨效果的测试结果图;
61.图23为实施例和对比例得到产品体内成骨效果的he染色结果图;
62.图24为实施例和对比例得到产品体内成骨效果的masson染色结果图;
63.图25为对比例4中plga film-pgc组膜进行扫描电镜(sem)图;
64.图26为对比例4中plga film-pgc组膜的元素分布图;
65.图27为plga film-pgc和plga film-pgn的拉伸应力应变曲线图;
66.图28为plga film-pgc和plga film-pgn的弹性模量图;
67.图29为plga film-pgc和plga film-pgn的断裂伸长率图;
68.图30为plga film-pgc和plga film-pgn膜表面亲水性测试结果图。
具体实施方式
69.为使本发明实施例的目的、技术方案和优点更加清楚,下面将对本发明实施例中的技术方案进行清楚、完整地描述。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
70.本发明实施例提供一种引导骨组织再生膜材料的制备方法,请参照图1,包括以下步骤:
71.s1、plga纤维膜的制备
72.plga(聚乳酸-羟基乙酸共聚物)纤维膜可以但不限于采用静电纺丝的方式制备,其制备过程包括:先制备plga静电纺丝溶液,采用静电纺丝的方法制备plga纤维膜。
73.在一些实施例中,在实际操作过程中plga纤维膜的制备过程包括:将plga颗粒和有机溶剂混合均匀后进行脱气处理得到plga静电纺丝溶液,将plga静电纺丝溶液注射至附着在接收滚筒上的接收膜材上形成静电纺丝纤维膜;控制纺丝电压为10-20kv,纺丝距离(纺丝距离是指注射器与接受滚筒之间的距离)为10-20cm,滚筒转速为700-900转/min,注射速率为3-5ml/h;纺丝期间温度控制在22-25℃,湿度为40%-60%。通过进一步调控静电纺丝的工艺参数,以得到更加均一的纤维膜产品。
74.具体地,控制纺丝电压为10kv、15kv、20kv等,纺丝距离可以为10cm、15cm、20cm等,滚筒转速可以为700转/min、800转/min、900转/min等,注射速率为3ml/h、4ml/h、5ml/h等,温度可以为22℃、23℃、24℃、25℃等,湿度可以为40%、50%、60%等。
75.具体地,可以采用一般的注射器连接21g(内径=0.5mm)的不锈钢金属喷头注射
plga静电纺丝溶液。
76.在一些实施例中,接收膜材选自锡箔、无纺布中的至少一种,以上几种材料均适合于作为静电纺丝过程中的接收膜材使用。
77.在一些实施例中,所使用的plga颗粒制备过程中控制聚乳酸和羟基乙酸的质量比为2-4:1,如可以为2:1、3:1、4:1等,是一种市购材料;plga静电纺丝溶液的浓度为0.1-0.3g/ml,如可以为0.1g/ml、0.2g/ml、0.3g/ml等。有机溶剂选自1,1,1,3,3,3-六氟异丙醇、n,n-二甲基甲酰胺和四氢呋喃中的至少一种,能够很好地溶解plga颗粒的有机溶剂理论上均可以采用,优选低毒性的溶剂。
78.在一些实施例中,将制备完成的plga纤维膜置于通风橱中30-60h(如30h、40h、50h、60h等),以去除残留的有毒溶剂。
79.s2、光固化水凝胶的制备
80.由聚乙二醇二丙烯酸酯、甲基丙烯酸酰化明胶和粘土制备得到光固化水凝胶,其混合步骤不限。
81.为保证水凝胶的均匀性,对混合步骤进行了优化:将聚乙二醇二丙烯酸酯和水混合得到pegda水凝胶,将pegda水凝胶与甲基丙烯酸酰化明胶颗粒混合得到pegda-gelma混合水凝胶,将pegda-gelma混合水凝胶与粘土混合后再与光引发剂混合。每一步混合时,均采用搅拌的手段保证混合的均匀度。
82.在一些实施例中,粘土为纳米粘土,粒径为20-100nm,使用纳米粘土粒径更加均一,更有利于制备得到性能均一的产品。
83.在一些实施例中,将pegda水凝胶与甲基丙烯酸酰化明胶颗粒混合,在40-80℃的条件下搅拌10-30min,搅拌转速为800-1500转/min,以保证制备得到均一的pegda-gelma混合水凝胶。
84.在一些实施例中,聚乙二醇二丙烯酸酯、甲基丙烯酸酰化明胶颗粒和粘土的质量比为10:0.5-1.5:1.0-2.0;每毫升水对应聚乙二醇二丙烯酸酯的用量为0.2-0.4g;每毫升水对应光引发剂的用量为0.0005-0.002g。通过对聚乙二醇二丙烯酸酯、甲基丙烯酸酰化明胶颗粒和粘土的用量进行优化,以进一步提升制备得到复合材料的力学性能,制备得到具有较高的空间维持性的gbr膜。
85.需要说明的是,光引发剂的种类不限,可以但不限于i2959,也可以为lap。
86.s3、光固化交联
87.将plga纤维膜浸泡在由聚乙二醇二丙烯酸酯、甲基丙烯酸酰化明胶和粘土制备得到的光固化水凝胶中,然后将附着有光固化水凝胶的plga纤维膜进行紫外照射,使光固化水凝胶交联固化,稳定附着在plga纤维膜的孔隙和表面。
88.在实际操作过程中,将plga纤维膜浸泡在光固化水凝胶中,待其完全浸透后从光固化水凝胶中取出,采用紫外灯在样品每侧均照射20-60s(如20s、30s、40s、50s、60s等)进行紫外光交联。通过短时间(如3-10秒)的浸泡就能使光固化水凝胶完全浸透plga纤维膜,取出后进行紫外光照射即可交联固化。
89.在一些实施例中,在浸泡之前,将plga纤维膜剪成所需形状。先根据最终需要的形状塑形,再光照就可以得到具有一定形状的gbr膜,在临床工作中一般遇到的骨缺损都是不规则形状的,可实现根据不同骨缺损而行个性化的gbr术。
90.本发明实施例还提供一种引导骨组织再生膜材料,其包括plga纤维膜,在plga纤维膜孔隙和表面均附着有水凝胶光固化材料;其中,水凝胶光固化材料的原料包括:聚乙二醇二丙烯酸酯、甲基丙烯酸酰化明胶和粘土。通过在plga纤维膜上附着由聚乙二醇二丙烯酸酯、甲基丙烯酸酰化明胶和粘土光固化形成的水凝胶光固化材料,制备得到的复合材料具备较高的空间维持性,具有较慢的降解性能,为缺损区的成骨提供足够的空间和时间条件。
91.在一些实施例中,聚乙二醇二丙烯酸酯、甲基丙烯酸酰化明胶和粘土的质量比为10:0.5-1.5:1.0-2.0;水凝胶光固化材料在引导骨组织再生膜材料中的质量占比为20-50%;plga纤维膜是通过静电纺丝的方式制备,其厚度为0.5-2mm。通过对材料的厚度、水凝胶光固化材料的含量进行优化,进一步提升其空间维持性,使降解时间较慢满足成骨的时间需求。
92.需要说明的是,本发明实施例制备得到的引导骨组织再生膜材料适合于进一步制备口腔骨缺损区修复材料,用于种植手术。
93.以下结合实施例对本发明的特征和性能作进一步的详细描述。
94.实施例1
95.本实施例提供一种引导骨组织再生膜材料的制备方法,包括以下步骤:
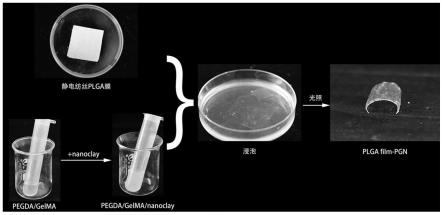
96.(1)plga静电纺丝膜的制备
97.将plga(聚乳酸和羟基乙酸的质量比为3:1)颗粒倒入盛有1,1,1,3,3,3-六氟异丙醇(hfip)的50ml离心管中,室温下在磁力搅拌器中搅拌过夜,以获得分散均匀地混合溶液,超声脱气15min,配成0.3g/ml的混合溶液作为静电纺丝溶液。
98.将上述静电纺丝溶液注入20ml的注射器中,连接21g(内径=0.5mm)不锈钢金属喷头,在15kv的纺丝电压和15cm的纺丝距离下,将锡箔纸粘贴在负电压相连的接收滚筒上,滚筒转速设置为800转/min,通过静电纺丝装置,用推注泵控制按照4ml/h的速度制备plga静电纺丝纤维膜,纺丝期间温度控制在22-25℃,湿度控制在40%-50%范围内,最后将制备完成的plga纤维(厚度为1.0mm)膜置于通风橱中48h,以去除残留的有毒溶剂。
99.(2)光固化水凝胶的制备
100.光固化水凝胶配置过程中所使用的所有玻璃器皿(烧杯、搅拌棒、玻璃培养皿和圆底烧瓶)均经高温高压灭菌后备用。粉末状材料(纳米粘土粉末、gelma冻干颗粒)分别以薄层平铺的形式放置于灭菌玻璃培养皿中,在肯格王紫外消毒柜内进行不少于24小时的紫外线照射消毒。溶剂及试剂(纯水、培养基、血清、双抗等)经0.22μm滤器过滤,纯水在过滤之后进行高温高压灭菌。磁力搅拌器、搅拌子、金属温度探头、金属称量勺等试验器材均通过高温高压蒸汽灭菌备用。配置过程均在超净工作台内完成,所有无法经高温消毒的设备均在超净台中经紫外线照射消毒至少24小时。
101.取干净的20ml圆底烧瓶,将3g pegda(即30%w/v)加入到10ml纯水中搅拌形成稳定溶液(即pegda水凝胶),再称量冻干gelma颗粒(300mg)加入pegda水凝胶中,60℃下用磁力搅拌器搅拌20min,搅拌速度1000转/min,即pegda/gelma水凝胶,最后加入400mg纳米粘土(粒径为20-100nm)(即4%w/v),继续搅拌形成稳定溶液,即可形成pegda/gelma/nanoclay水凝胶,最终在四组水凝胶中均加入10mg光引发剂i2959(即0.1%w/v),避光搅拌10min,即可制成所需的各组光固化水凝胶,该水凝胶制备后即可使用或放置在-4℃冰箱中
避光保存。
102.(3)制备gbr膜
103.将静电纺丝制成的plga膜浸泡在光固化水凝胶中,浸泡前将plga纤维膜剪成所需要的形状,待其完全浸透后从溶液中取出plga膜,去除膜表面多余的水凝胶,使用紫外灯在样品每侧照射30s进行紫外光交联。光交联后,将样品置于4℃的冰箱中保存,室温下于真空烘箱中干燥,记为plga film-pgn。
104.实施例2
105.本实施例提供一种引导骨组织再生膜材料的制备方法,包括以下步骤:
106.(1)plga静电纺丝膜的制备
107.将plga(聚乳酸和羟基乙酸的质量比为2:1)颗粒倒入盛有1,1,1,3,3,3-六氟异丙醇(hfip)的50ml离心管中,室温下在磁力搅拌器中搅拌过夜,以获得分散均匀地混合溶液,超声脱气15min,配成0.3g/ml的混合溶液作为静电纺丝溶液。
108.将上述静电纺丝溶液注入20ml的注射器中,连接21g(内径=0.5mm)不锈钢金属喷头,在10kv的纺丝电压和10cm的纺丝距离下,将锡箔纸粘贴在负电压相连的接收滚筒上,滚筒转速设置为700转/min,通过静电纺丝装置,用推注泵控制按照3ml/h的速度制备plga静电纺丝纤维膜,纺丝期间温度控制在22-25℃,湿度控制在40%-50%范围内,最后将制备完成的plga纤维膜(厚度为0.5mm)置于通风橱中30h,以去除残留的有毒溶剂。
109.(2)光固化水凝胶的制备
110.取干净的20ml圆底烧瓶,将2g pegda(即20%w/v)加入到10ml纯水中搅拌形成稳定溶液(即pegda水凝胶),再称量冻干gelma颗粒(100mg)加入pegda水凝胶中,40℃下用磁力搅拌器搅拌30min,搅拌速度800转/min,即pegda/gelma水凝胶,最后加入200mg纳米粘土(粒径为20-100nm)(即4%w/v),继续搅拌形成稳定溶液,即可形成pegda/gelma/nanoclay水凝胶,最终在四组水凝胶中均加入5mg光引发剂i2959(即0.1%w/v),避光搅拌10min,即可制成所需的各组光固化水凝胶,该水凝胶制备后即可使用或放置在-4℃冰箱中避光保存。
111.(3)制备gbr膜
112.将静电纺丝制成的plga膜浸泡在光固化水凝胶中,浸泡前将plga纤维膜剪成所需要的形状,待其完全浸透后从溶液中取出plga膜,去除膜表面多余的水凝胶,使用紫外灯在样品每侧照射20s进行紫外光交联。光交联后,将样品置于4℃的冰箱中保存,室温下于真空烘箱中干燥,记为plga film-pgn。
113.实施例3
114.本实施例提供一种引导骨组织再生膜材料的制备方法,包括以下步骤:
115.(1)plga静电纺丝膜的制备
116.将plga(聚乳酸和羟基乙酸的质量比为4:1)颗粒倒入盛有1,1,1,3,3,3-六氟异丙醇(hfip)的50ml离心管中,室温下在磁力搅拌器中搅拌过夜,以获得分散均匀地混合溶液,超声脱气15min,配成0.3g/ml的混合溶液作为静电纺丝溶液。
117.将上述静电纺丝溶液注入20ml的注射器中,连接21g(内径=0.5mm)不锈钢金属喷头,在20kv的纺丝电压和20cm的纺丝距离下,将锡箔纸粘贴在负电压相连的接收滚筒上,滚筒转速设置为900转/min,通过静电纺丝装置,用推注泵控制按照5ml/h的速度制备plga静
电纺丝纤维膜,纺丝期间温度控制在22-25℃,湿度控制在50%-60%范围内,最后将制备完成的plga纤维膜(厚度为2.0mm)置于通风橱中60h,以去除残留的有毒溶剂。
118.(2)光固化水凝胶的制备
119.取干净的20ml圆底烧瓶,将4g pegda(即30%w/v)加入到10ml纯水中搅拌形成稳定溶液(即pegda水凝胶),再称量冻干gelma颗粒(600mg)加入pegda水凝胶中,80℃下用磁力搅拌器搅拌10min,搅拌速度1500转/min,即pegda/gelma水凝胶,最后加入800mg纳米粘土(粒径为20-100nm)(即4%w/v),继续搅拌形成稳定溶液,即可形成pegda/gelma/nanoclay水凝胶,最终在四组水凝胶中均加入20mg光引发剂i2959(即0.1%w/v),避光搅拌10min,即可制成所需的各组光固化水凝胶,该水凝胶制备后即可使用或放置在-4℃冰箱中避光保存。
120.(3)制备gbr膜
121.将静电纺丝制成的plga膜浸泡在光固化水凝胶中,浸泡前将plga纤维膜剪成所需要的形状,待其完全浸透后从溶液中取出plga膜,去除膜表面多余的水凝胶,使用紫外灯在样品每侧照射60s进行紫外光交联。光交联后,将样品置于4℃的冰箱中保存,室温下于真空烘箱中干燥,记为plga film-pgn。
122.对比例1
123.与实施例1不同之处仅在于:不进行步骤(2)和步骤(3),记为plga film。
124.对比例2
125.与实施例1不同之处仅在于:步骤(2)中不加入gelma颗粒和纳米粘土,记为plga film-p。
126.对比例3
127.与实施例1不同之处仅在于:步骤(2)中不加入纳米粘土,记为plga film-pg。
128.对比例4
129.与实施例1不同之处仅在于:将纳米粘土替换为等量的纳米纤维素,记为plga film-pgc。
130.试验例1
131.观察静电纺丝plga膜,应为白色、无污染、表面由交叉的纤维丝组成,该膜浸泡入光固化水凝胶中后,应为透明、表面光滑的柔软膜,光固化后的膜应为透明、具有较高机械强度的光滑膜。对实施例和对比例制备得到的各组膜层进行性能表征如下:
132.(1)对各组膜进行扫描电镜(sem)观察表面形貌及元素分布图,结果如图2所示
133.从图2可以看出,plga film(a)由无序的纳米纤维组成,plga film-p(b)、plga film-pg(c)、plga film-pgn(d)表面粗糙,这四种膜的表面形态均有利于细胞的粘附与增殖,由eds结果可知,四种膜的c、n、o三种元素均匀分布,说明各组分在膜中均匀分布。
134.(2)利用同步热分析测定各组gbr膜的不同温度、时间下不同材料的交联程度,研究各组材料的交联结构、分子结构,热重测试结果图如图3所示。
135.由热重结果可以得出四组膜的曲线形状没有差异,都是首先发生快速的重量减轻(脱水),然后曲线变的平缓,最终再发生重量的明显减轻。
136.(3)利用接触角测量仪测定各组gbr膜表面亲水性,测定各组gbr膜的平衡溶胀率测定,结果如图4所示。
137.从图4可以看出,实验结果得各组膜的表面接触角均为锐角,可见各组膜有着很强的亲水性。其中plga film-pgn的接触角最小,说明其亲水性最好。
138.(4)在体外降解实验中,将gbr膜浸泡在pbs和溶菌酶溶液中(37℃,水平摇床),每周取出后测量材料失重率和pbs浸泡液ph变化,结果如图5所示。
139.从图5可以看出,各组膜在37℃时1h的平衡溶胀率都保持在50%-60%之间,并且各组之间差异不存在统计学意义,说明各组膜均有一定的溶胀率。
140.(5)对各组膜进行体外降解试验,将gbr膜置入大鼠颅部皮下1个月后取出,分析其体内降解性,测试结果如图6、图7和图8所示。
141.根据图6-图8可知,根据不同时间的降解测量数据分析折线图可得,不同时间各组的降解量基本上随着时间的增加而逐渐增大,其中plga膜在pbs和溶菌酶溶液中均降解的最快,其他三组膜降解缓慢;plga膜的ph值4周后下降速率增快,主要是因为plga分解后产生酸性的降解产物,其余三组的ph保持在6.5左右,说明水凝胶中的降解产物可以中和plga的酸性降解产物。
142.(6)利用万能力学测试仪检测各组gbr膜的机械强度及其断裂伸长率,结果如图9、图10、图11所示。
143.从图9-图11可知,plga film-pgn具有较高的拉伸模量和较低的伸长率,说明其强度较高。
144.(7)如图12所示,将plga film-pgn膜在光照前如图塑性(圆拱形),光照后其可以承受较大的外力(50g)而不发生形变,说明plga film-pgn具有较高的空间维持性和可塑形性。
145.(8)细胞cck-8实验(bmsc、l929)
146.试验方法:cck-8细胞毒性实验检测各组新型gbr膜对bmsc细胞和l929细胞的毒性;结果如图13和图14所示。
147.结果表明,各组膜均有利于bmsc和l929细胞增殖,其中在plga film-pgn上的细胞具有最高的活性。
148.(9)测试bmsc细胞在gbr膜上的粘附和活死,结果如图15和图16所示。
149.测试方法:荧光电镜(1天和3天)
150.由图15和图16可知,plga film-pgn具有良好的理化性能,其力学强度也符合gbr膜的要求,并且有利于细胞的增殖与粘附,因此以下的实验中将不再把同由plga膜和光固化水凝胶组成的plga film-p和plga film-pg这两组纳入实验组,而是保留plga film和plga film-pgn两个实验组与对照组进行实验对比。
151.(10)bmsc细胞在gbr膜上的成骨分化测试,结果如图17、图18、图19和图20所示。
152.测试方法:光镜观察
153.通过alp、茜素红的定性定量实验可以得出,plga film-pgn有利于bmsc细胞的成骨分化。
154.(11)通过l929细胞屏障实验观察gbr膜的屏障功能,结果如图21所示。
155.测试方法:光镜观察(结晶紫染色)
156.通过图21可以看出,plga film-pgn通过gbr膜的l929细胞最少,说明其有一定的细胞屏障作用。
157.(12)体内成骨效果
158.将gbr膜置入大鼠颅部皮下1个月后取出,分析其体内降解性,并且通过五脏he切片分析体内毒性;在大鼠颅部造5mm圆形骨缺损,将gbr膜置入后1个月,通过micro-ct、he染色、masson染色观察其体内成骨效果,如图22、图23和图24所示。
159.在动物实验中引入了一个阳性对照组:海奥膜(目前临床常用gbr膜),micro-ct结果可以看出plga film-pgn的新生骨最多,he和masson染色也可以看出plga film-pgn新生骨最多,骨缺损区域缩小,其中新生骨具有一定板层结构,缺损区的纤维结缔组织较少,说明该膜具有一定的成骨活性和细胞屏障作用,符合gbr膜的要求。
160.(13)对比例4中plga film-pgc组膜进行扫描电镜(sem)观察表面形貌及元素分布图,结果如图25和图26所示。
161.从图25可以看出,plga film-pgc表面粗糙,纳米纤维素在膜的表面分布不均匀,由图26的元素分布结果可知,四种膜的c、n、o三种元素均匀分布。
162.利用万能力学测试仪检测plga film-pgc和plga film-pgn的拉伸应力应变曲线、弹性模量及其断裂伸长率,结果如图27、图28、图29所示,由结果显示,添加纳米纤维素不能有效的提高其机械强度。
163.利用接触角测量仪测定plga film-pgc和plga film-pgn膜表面亲水性,结果如图30所示。
164.从图30可以看出,实验结果得三组膜的表面接触角均为锐角,但是,plga film-pgc的角度大于plga film和plga film-pgn,表明其亲水性没有plga film-pgn好。
165.综上,对比例4中的plga film-pgc膜的纳米纤维素分布、机械强度以及亲水性均没有实验组中的plga film-pgn理想。
166.综上,本发明提供一种引导骨组织再生膜材料的制备方法,结合静电纺丝和光固化水凝胶来实现gbr膜的制备的,主要提高机械强度的成分是pegda和纳米粘土,具有以下优点:
167.(1)该gbr膜具有较高的空间维持性,避免患者术后在咀嚼和说话过程骨缺损区中受到唇颊肌的压力而塌陷,为缺损区的成骨提供足够的空间和时间条件。口腔gbr术后需要gbr膜长时间维持一定的成骨空间,如果膜的机械强度不够,则会影响成骨效果,本发明中的plga film-pgn膜具有很高的机械强度,拉伸模量可高达130mpa,具有一定的空间维持性。
168.(2)该gbr膜具有较慢的降解性能,如果降解太快,则无法起到屏障作用不利于成骨,该gbr膜能够实现长时间的屏障作用,有利于防止非成骨细胞和组织进入骨缺损区而影响成骨效果。
169.(3)该gbr膜中的成分均有利于细胞的增殖和成骨分化,并且具有一定的细胞屏障作用,符合临床gbr膜的要求。
170.(4)plga film-pgn具有较低的溶胀,避免早期gbr术创口开裂,并且具有降解可调性,能够将长时间的维持成骨所需的空间并且防止非成骨细胞进入缺损区。
171.以上仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1