一种利用单核苷酸多态性判断拷贝数变异的方法和系统与流程
1.本发明涉及一种判断拷贝数变异的方法和一种数据处理系统,特别涉及一种利用单核苷酸多态性判断拷贝数变异的方法和一种实施该方法的数据处理系统。
背景技术:
2.随着个体化医学的发展和"精准医学"概念的提出,肿瘤药物治疗发展迅速,临床研究逐渐发现并证实更多与药物治疗疗效预测相关的基因突变。传统的基因突变检测方法如sanger测序、焦磷酸测序和实时荧光pcr等仅能对单个基因,或者单个基因的部分外显子突变进行检测,采用上述传统基因突变检测方法同时检测多个基因,一则需要的样本量大,其次需要更长的检测时间和更大的工作量。高通量测序(high-throughput sequencing,hts)即下一代测序(next-generation sequencing technology,ngs),能够同时对上百万甚至数十亿个dna片段进行测序,可实现在较低的成本下,一次对多至上百个肿瘤相关基因、全外显子以及全基因组进行检测,而且需要的样本量并不增加。因其在通量、成本和效率方面的优势,ngs在实体肿瘤体细胞基因突变中展现了其广阔的应用前景。
3.拷贝数变异在人类疾病和生物学中起着重要作用。例如,生殖细胞拷贝数变异会对发育有巨大影响,如21、18和13三体,分别导致唐氏综合征、爱德华兹综合征和帕托综合征等。另一方面,在患癌患者中常常会发现体细胞拷贝数变异,拷贝数变异是肿瘤发展和耐药性的主要驱动因素。泛癌基因组分析报告中发现在许多肿瘤类型中发现了myc的拷贝数扩增和pten和tp53的拷贝数缺失。在急性髓系白血病(aml)中,涉及大量染色体5和7的缺失在细胞遗传风险不利的患者中经常出现。在多发性骨髓瘤中,17号染色体的缺失与更侵袭性的疾病相关,在疾病进展过程中获得17号染色体的缺失会带来更糟糕的预后。此外,tp53染色体缺失或1号染色体扩增导致骨髓瘤发病相关基因(如cks1b、mcl1)异常,与不良预后相关。因此,对癌症相关拷贝数变异事件的描述对于确定患者亚群以及对预后和潜在治疗策略的见解有重要意义。
4.目前ngs方法检测cnv主要可利用1)、读段深度(read depth):根据滑动窗口读段深度来指示拷贝数扩增与缺失;2)、pair-end方法:根据pair-end两端之间距离与参考基因组上差异来确认拷贝数变异;3)、序列组装方法:将短reads进行组装后寻找其与参考基因组之间的差异来确认拷贝数变异。第一种基于读段深度的方法是目前应用较为广泛的方法,后两种主要被用于进行其他结构变异的检测,如转换颠换等。读段深度检测方法的核心技术主要基于概率统计模型。基于概率统计的检测方法有一个假设前提:读段深度与拷贝数变异数目之间是线性关系,即我们默认测序过程是均匀的,染色体上按特定窗口进行滑动统计的读段深度是服从某种特定分布的,比如泊松分布、高斯分布等。如果出现滑动窗口读段深度增加或者减少也就代表着出现拷贝数的扩增或者缺失。但是测序过程中累积的误差使得读段深度与拷贝数变异数目之间并非是线性关系,因此该方法基于错误的假设,得出的结果误差较大。
5.此外,也可以依据细胞标志基因,通过人工鉴别,该方法效率低下,且存在较多的
主观性。
6.为解决以上方法的局限性,开发了一种方法基于基因范围中诸多单核苷酸多态性的频率来辅助确定基因拷贝数变异的状态。
技术实现要素:
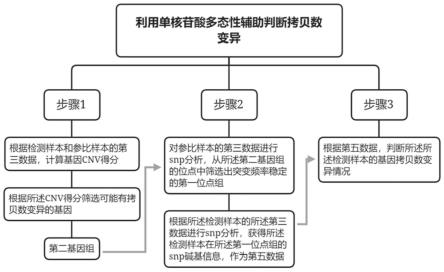
7.为解决上述问题,本发明提供一种利用单核苷酸多态性辅助判断拷贝数变异的方法,其包括以下步骤:
8.步骤1:根据检测样本和参比样本的第三数据,计算基因cnv得分,根据所述cnv得分筛选可能有拷贝数变异的基因,选入第二基因组;
9.步骤2:根据所述参比样本的所述第三数据进行snp分析,从所述第二基因组的位点中筛选出突变频率稳定的第一位点组;根据所述检测样本的所述第三数据进行snp分析,获得所述检测样本在所述第一位点组的snp碱基信息,作为第五数据;
10.步骤3:根据第五数据,判断所述所述检测样本的基因拷贝数变异情况;
11.所述第三数据是原始测序数据经过去除低质量序列、比对到参考基因组、去重和质控后获得的数据。
12.所述低质量序列是指平均碱基质量和reads长度低于设置数值的序列,本发明实施例一中给出了示例性的低质量序列去除方式。
13.本发明可以用于第二代测序,或称高通量测序平台(例如illumina或者mgi测序数据)的测序结果进行第一基因组的拷贝数变异状态的识别。
14.所述第三数据是通过如下方法获取的:
15.针对检测样本或参比样本进行测序,获得包括第一基因组信息的原始测序数据,所述第一基因组是根据分析目的自行选定的一个或多个基因;
16.所述原始测序数据去除低质量序列得到的clean data作为第一数据,将所述第一数据比对到参考基因组,从而将第一数据中的测序序列定位到相关基因上,得到比对结果数据,作为第二数据;
17.对所述第二数据进行去重和质控,得到第三数据。
18.比对到参考基因组的方法为:使用bwa软件,利用mem算法,将去除低质量序列得到的第一数据比对到人参考基因组hg19上,从而将测序序列定位到相关基因上。
19.参比样本选用人白细胞样本,为保证样本的多样性和结果具有统计学意义,可以选取多人的血液样本,本发明中选用20人的血液样本构建cnv和snp基线。
20.作为优选的方案,步骤1具体包括以下步骤:
21.步骤1.1获取检测样本和参比样本的校正的标准化测序深度;
22.使用多份参比样本时,对所有参比样本的校正的标准化测序深度取平均值,计算平均参比测序深度;
23.步骤1.2计算检测样本的cnv拷贝数得分;计算方式为:对于每一个区域,该区域的cnv拷贝数得分=2
×
校正的标准化测序深度/平均参比测序深度;
24.步骤1.3筛选0≤cnv拷贝数得分≤1,或者4≤cnv拷贝数得分<6的样本,作为可能有拷贝数变异的基因。
25.作为优选的方案,步骤2具体包括以下步骤:
26.步骤2.1从所述参比样本的所述第三数据中获取所述第二基因组的snp位点的碱基信息,包括支持碱基突变的reads和参考碱基的reads;去除其中测序深度低于设定值的位点,得到的数据称为参比样本的第四数据;
27.利用参比样本的第四数据进行如下计算和筛选:
28.snp突变频率=支持碱基突变的reads数/(参考碱基的reads数+支持碱基突变的reads数);
29.筛选的标准为,snp位点突变频率在0.4-0.6之间则判定为稳定,归为第一位点组。
30.步骤2.2从所述检测样本的所述第三数据中获取所述第一位点组的snp位点的碱基信息,包括支持碱基突变的reads和参考碱基的reads;去除其中测序深度低于设定值的位点,得到的数据称为参比样本的第五数据。
31.步骤3.1利用检测样本的第五数据进行如下计算:
32.snp突变频率=支持碱基突变的reads数/(参考碱基的reads数+支持碱基突变的reads数);
33.步骤3.2,如果snp的分布频率<0.3时表示拷贝数可能发生缺失,基因标准化校正后的snp的分布频率>0.7时表示拷贝数可能发生扩增。
34.本发明进一步提供一种利用单核苷酸多态性判断拷贝数变异的系统,其包括:
35.存储器,所述存储器存储可执行指令,以及;以及
36.一个或多个处理器,所述一个或多个处理器与所述存储器通信以执行可执行指令从而完成以下操作:
37.步骤1:根据检测样本和参比样本的第三数据,计算基因cnv得分,根据所述cnv得分筛选可能有拷贝数变异的基因,选入第二基因组;
38.步骤2:根据所述参比样本的所述第三数据进行snp分析,从所述第二基因组的位点中筛选出突变频率稳定的第一位点组;根据所述检测样本的所述第三数据进行snp分析,获得所述检测样本在所述第一位点组的snp突变频率数据,作为第五数据;
39.步骤3:根据第五数据,判断所述检测样本的所述第二基因组中基因的拷贝数变异情况;
40.所述第三数据是原始测序数据经过去除低质量序列、比对到参考基因组、去重和质控后获得的数据。
41.作为优选,所述一个或多个处理器与所述存储器通信以执行可执行指令从而完成以下操作:
42.步骤1.1获取检测样本和参比样本的校正的标准化测序深度;
43.使用多份参比样本时,对所有参比样本的校正的标准化测序深度取平均值,计算平均参比测序深度;
44.步骤1.2计算检测样本的cnv拷贝数得分;
45.计算方式为:对于每一个区域,该区域的cnv拷贝数得分=2
×
校正的标准化测序深度/平均参比测序深度;
46.步骤1.3筛选0≤cnv拷贝数得分≤1,或者4≤cnv拷贝数得分<6的样本,作为可能有拷贝数变异的基因;
47.步骤2.1从所述参比样本的所述第三数据中获取所述第二基因组的snp位点的碱
基信息,包括支持碱基突变的reads和参考碱基的reads;去除其中测序深度低于设定值的位点,得到的数据称为参比样本的第四数据;
48.利用参比样本的第四数据进行如下计算和筛选:
49.snp突变频率=支持碱基突变的reads数/(参考碱基的reads数+支持碱基突变的reads数);
50.筛选的标准为,snp位点突变频率在0.4-0.6之间则判定为稳定,归为第一位点组;
51.步骤2.2从所述检测样本的所述第三数据中获取所述第一位点组的snp位点的碱基信息,包括支持碱基突变的reads和参考碱基的reads;去除其中测序深度低于设定值的位点,得到的数据称为参比样本的第五数据;
52.步骤3.1利用检测样本的第五数据进行如下计算:
53.snp突变频率=支持碱基突变的reads数/(参考碱基的reads数+支持碱基突变的reads数);
54.步骤3.2,如果snp的分布频率<0.3时表示拷贝数可能发生缺失,基因标准化校正后的snp的分布频率>0.7时表示拷贝数可能发生扩增。
55.本发明的优点在于:
56.采用snp分布频率辅助判断基因拷贝数变异情况,相比传统单纯依靠测序深度判断,结果更准确。
57.在优选的方案中,能够排除测序数据质量低、深度不足等带来的误差。
附图说明
58.通过阅读参照以下附图所作的对非限制性实施例所作的详细描述,本技术的其它特征、目的和优点将会变得更明显:
59.图1为实施例一基因cnv得分分布图;
60.图2为实施例一参比样本(白细胞)snp突变频率基线图;
61.图3为实施例一检测样本第一位点组的snp突变频率分布图;
62.图4为本发明技术方案实施流程图;
63.图5为本发明实施例二所示的数据处理系统示意图。
具体实施方式
64.下面结合附图和实施例对本技术作进一步的详细说明。可以理解的是,此处所描述的具体实施例仅仅用于解释相关发明,而非对该发明的限定。另外还需要说明的是,为了便于描述,附图中仅示出了与有关发明相关的部分。
65.需要说明的是,在不冲突的情况下,本技术中的实施例及实施例中的特征可以相互组合。下面将参考附图并结合实施例来详细说明本技术。
66.实施例一
67.本实施例中,选定肿瘤相关基因作为第一基因组。
68.选定广东某医院送检肿瘤样本作为检测样本;选定分别来源于20个健康成年人的20份血液样本中提取的白细胞样本作为参比样本。
69.测序和测序数据的前期处理可使用常规的方法和软件完成,本实施例提供示例性
的操作如下:
70.采用illumina二代测序平台,对检测样本和参比样本测序,获得原始测序数据。
71.对检测样本的原始测序数据进行前期处理:a.原始数据的低质量序列的去除:因为测序仪的原因一般测序数据在测序起始和测序结束阶段碱基质量会出现波动和测序质量较低的情况,如果插入片段较短时也存在测序测通,即测序测到包含测序接头的情况,因此为了确保样本分析结果的准确性,需要将测序质量较低的序列或者测序引入的测序接头进行去除。去除标准如下:去除测序使用的illumina接头,以4bp为滑窗去除平均碱基质量低于15的碱基,去掉长度低于26的reads,从而得到clean data,为方便表述,以下称为第一数据。
72.b.比对参考基因组:利用bwa算法,将去除完低质量序列的clean data序列比对到人参考基因组上,从而将测序序列定位到相关基因上,从而得到第二数据。
73.c.比对结果去重:因为实验过程中存在pcr扩增,为了避免扩增数据带来的影响,所以使用picard软件的markduplicates模块对比对上的数据(即第二数据)进行去重。
74.d.样本测序质量的质控:基于比对的结果对样本的测序质量和比对质量进行评估,从而判断样本质量是否满足要求,对于不能满足要求的样本无法确保分析结果的准确性。对去重后的数据进行质控,质控标准如下:在靶率》60%,q30》85%,比对率》95%,去重后平均深度》1000x。第二数据经过去重和质控,得到第三数据。
75.对每一份参比样本的原始测序数据也进行同样处理,得到每份参比样本的第三数据。
76.步骤1
77.基因cnv得分的计算和筛选:
78.1.1获取校正的标准化测序深度
79.利用bedtools软件的multicov功能来计算样本的第三数据在第一基因组区域内各基因的测序深度;计算出整个样本在第一基因组区域中的总测序深度;根据每个基因的测序深度和计算获得的总测序深度进行标准化;使用loess方法基于gc含量对标准化之后的测序深度结果进行校正。
80.对参比样本,采用相同方式处理数据,在获得每个参比样本的处理结果之后取平均值,得到平均参比测序深度。
81.1.2计算检测样本的cnv拷贝数得分
82.计算方式为:对于每一个区域,该区域的cnv拷贝数得分=2
×
校正的标准化测序深度/平均参比测序深度;
83.1.3筛选出可能发生拷贝数变异的基因
84.所述筛选标准为:基因cnv拷贝数得分≥4,且<6,则可能发生扩增;基因cnv得分≤1则可能发生缺失;以上两种情况的基因均选入第二基因组。
85.参见说明书附图1,图中示例性地标出的egfr,met,erbb2基因的cnv拷贝数得分符合筛选标准,有较大可能发生了拷贝数变异。
86.步骤2
87.snp数据的处理,具体地,包括:
88.2.1筛选snp突变频率稳定的位点
89.通过20个白细胞样本的snp位点突变频率的分析汇总,从中筛选出第二基因组中突变频率稳定的位点。
90.具体地,利用bcftools软件的mpileup功能,从每个参比样本的第三数据中获取第二基因组的snp位点的碱基信息;将其作为输入,用r语言进行读取,能够获取样本中第二基因组中所有相关的reads,包括支持碱基突变的reads和参考碱基的reads。对于每个snp位点,如果支持碱基突变的reads数+参考碱基的reads数<200,证明测序深度不够,可能影响分析结果的准确性,需要去除。经过上述处理后得到的数据称为参比样本的第四数据。
91.利用参比样本的第四数据进行如下计算和筛选:
92.snp突变频率=支持碱基突变的reads数/(参考碱基的reads数+支持碱基突变的reads数)
93.筛选的标准为,snp位点突变频率在0.4-0.6之间则判定为稳定,归为第一位点组。
94.为直观显示结果,可以作白细胞snp突变频率基线,参见说明书附图2。
95.在白细胞中突变频率不稳定的snp位点,在其他细胞中一般也表现为不稳定,无法用本发明的方法对其进行分析,去除该部分位点的数据,可以避免部分snp位点异常导致的结果错误。
96.2.2获取检测样本的第五数据
97.从所述检测样本的所述第三数据中获取所述第一位点组的snp位点的碱基信息,包括支持碱基突变的reads和参考碱基的reads;去除其中测序深度低于设定值的位点,得到的数据称为参比样本的第五数据。
98.具体地,利用bcftools软件的mpileup功能,从检测样本的第三数据中获取第一位点组的snp位点的碱基信息;对于每个snp位点,如果支持碱基突变的reads数+参考碱基的reads数<200,证明测序深度不够,可能影响分析结果的准确性,需要去除。经过上述处理后得到的数据称为检测样本的第五数据。
99.步骤3拷贝数变异的判断。
100.3.1检测样本snp突变频率的计算
101.利用检测样本的第五数据进行如下计算:
102.snp突变频率=支持碱基突变的reads数/(参考碱基的reads数+支持碱基突变的reads数);
103.3.2拷贝数变异判断
104.根据步骤3.1计算得到的snp突变频率数据,筛选snp突变频率<0.3的基因作为可能发生拷贝数缺失的第四基因组,筛选snp突变频率>0.7的基因作为可能发生拷贝数扩增的第五基因组。
105.为直观显示,可以利用计算获得的snp突变频率数据进行绘图,如说明书附图3所示,从图3中明显可见不同基因可能发生拷贝数变异的位点差距。cnv未发生缺失和突变的情况下,snp突变频率的分布一般在0.4-0.6之间,与白细胞snp基线接近。cdkn2a和cdkn2b基因有拷贝数缺失的可能,erbb2和met基因则有拷贝数扩增的可能。
106.作为本发明技术方案的优点的一种示例,根据图1判断,很可能发生拷贝数变异的egfr基因,从图3来看基本可以排除拷贝数变异的可能。这可以证明本发明使用snp辅助判断比现有技术具有更高的准确性。
107.需要说明的是,绘图的意义在于更直观地展示数据,即使不进行绘图,也可以通过数据处理完成本发明的技术方案。
108.实施例二
109.本技术还提供了一种利用单核苷酸多态性判断拷贝数变异的系统,可以通过移动终端、个人计算机(pc)、平板电脑、服务器等形式实现。下面参考图5,其示出了适于用来实现本技术实施方式的利用单核苷酸多态性判断拷贝数变异的系统的结构示意图。
110.如图5所示,计算机系统300包括一个或多个处理器、通信部等,所述一个或多个处理器例如:一个或多个中央处理单元(cpu)301,和/或一个或多个图像处理器(gpu)313等,处理器可以根据存储在只读存储器(rom)302中的可执行指令或者从存储部308加载到随机存取存储器(ram)303中的可执行指令而执行各种适当的动作和处理。通信部312可包括但不限于网卡,所述网卡可包括但不限于ib(infiniband)网卡。
111.处理器可与只读存储器302和/或随机存取存储器303通信以执行可执行指令,通过总线304与通信部312相连、并经通信部312与其他目标设备通信,从而完成本技术实施方式提供的任一项方法对应的操作,例如:
112.步骤1.2计算检测样本的cnv拷贝数得分;
113.计算方式为:对于每一个区域,该区域的cnv拷贝数得分=2
×
校正的标准化测序深度/平均参比测序深度;
114.步骤1.3筛选0≤cnv拷贝数得分≤1,或者4≤cnv拷贝数得分<6的样本,作为可能有拷贝数变异的基因;
115.步骤2.1从所述参比样本的所述第三数据中获取所述第二基因组的snp位点的碱基信息,包括支持碱基突变的reads和参考碱基的reads;去除其中测序深度低于设定值的位点,得到的数据称为参比样本的第四数据;
116.所述测序深度的设定值是操作人员根据测序时扩增次数等可以自行调整的,目的是减少干扰,提高分析的准确度。本实施例中,步骤2.1和2.2均设定为200。
117.利用参比样本的第四数据进行如下计算和筛选:
118.snp突变频率=支持碱基突变的reads数/(参考碱基的reads数+支持碱基突变的reads数);
119.筛选的标准为,snp位点突变频率在0.4-0.6之间则判定为稳定,归为第一位点组;
120.步骤2.2从所述检测样本的所述第三数据中获取所述第一位点组的snp位点的碱基信息,包括支持碱基突变的reads和参考碱基的reads;去除其中测序深度低于设定值的位点,得到的数据称为参比样本的第五数据;
121.步骤3.1利用检测样本的第五数据进行如下计算:
122.snp突变频率=支持碱基突变的reads数/(参考碱基的reads数+支持碱基突变的reads数);
123.步骤3.2,如果snp的分布频率<0.3时表示拷贝数可能发生缺失,基因标准化校正后的snp的分布频率>0.7时表示拷贝数可能发生扩增。
124.此外,在ram 303中,还可存储有装置操作所需的各种程序和数据。cpu 301、rom 302以及ram 303通过总线304彼此相连。在有ram 303的情况下,rom 302为可选模块。ram 303存储可执行指令,或在运行时向rom 302中写入可执行指令,可执行指令使处理器301执
行上述通信方法对应的操作。输入/输出接口(i/o接口)305也连接至总线304。通信部312可以集成设置,也可以设置为具有多个子模块(例如多个ib网卡),并在总线链接上。
125.以下部件连接至i/o接口305:包括键盘、鼠标等的输入部306;包括诸如阴极射线管(crt)、液晶显示器(lcd)等以及扬声器等的输出部307;包括硬盘等的存储部308;以及包括诸如lan卡、调制解调器等的网络接口卡的通讯部309。通讯部309经由诸如因特网的网络执行通信处理。驱动器310也根据需要连接至i/o接口305。可拆卸介质311,诸如磁盘、光盘、磁光盘、半导体存储器等等,根据需要安装在驱动器310上。
126.需要说明的,如图5所示的架构仅为一种可选实现方式,在具体实践过程中,可根据实际需要对上述图5的部件数量和类型进行选择、删减、增加或替换;在不同功能部件设置上,也可采用分离设置或集成设置等实现方式,例如gpu和cpu可分离设置或者可将gpu集成在cpu上,通信部312可分离设置,也可集成设置在cpu或gpu上,等等。这些可替换的实施方式均落入本发明公开的保护范围。
127.特别地,根据本技术,参考流程图5描述的过程可以被实现为计算机程序产品。例如,本技术提供一种计算机程序产品,包括计算机可读指令,所述计算机可读指令被处理器执行时实现以下操作:在这样的实施方式中,该计算机程序产品可以通过通讯部309从网络上被下载和安装,和/或从可拆卸介质311中读取并安装。在该计算机程序产品被中央处理单元(cpu)301执行时,执行本技术的方法中限定的上述功能。
128.可能以许多方式来实现本技术的技术方案。例如,可通过软件、硬件、固件或者软件、硬件、固件的任何组合来实现本技术的技术方案。用于说明方法的步骤顺序仅是为了更清楚地说明技术方案的目的而提供。除非经特别限定,否则本技术的方法步骤不限于以上具体描述的顺序。此外,在一些实施方式中,还可将本技术实施为存储计算机程序产品的存储介质。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1