恶性肺结节筛查基因标志物、筛查模型的构建方法和检测装置与流程
本发明涉及对影像表现为高风险的肺结节(radiographically highlysuspicious lung nodules)良恶性早筛, 属于分子生物医学。
背景技术:
1、肺癌是其中一个世界上确证率最高的癌症,高风险人群包括65岁以上,具有下列一项以上的危险因素者。危险因素包括:重度吸烟,曾经拥有抽烟史,家庭病史,接受过胸腔放射性治疗,致癌物质。由于肺癌前期缺乏明显症状,患者一般于肺癌中后期(stage iii,iv)被诊断出。然而,大量研究表明在早期被诊断出的肺癌患者能有更高的生存率。在肺癌一期(stage i)被诊断出的患者比在肺癌四期(stage iv)被诊断出的患者的五年存活率提高13倍。因此,肺部肿瘤早发现和早诊断对于提高肺癌患者的生存率至关重要。
2、低剂量胸腔电脑断层扫描(low-dose computed tomography thorax, ldct)检测肺结节是现今发现肺部肿瘤最常见的诊断方式。针对影像学判定的肺结节进行手术切除,能有效地减少20%-39% 的肺癌死亡率。然而,大约15%-35% 的肺结节,在初始ldct影像表现判定为高风险的肺结节,在手术切除后,最后被确认为病理性无害。因此,影像学检测存在一定的局限性,仅依据影像检测结果进行恶性肺肿瘤诊断,增加了一些非必要的手术,造成患者非必要的手术风险和并发症风险,和增加了医疗支出负担。因此,对仅在影像学依据下判定为肺癌高危人群的肺结节良恶性判断工作尤为重要。
技术实现思路
1、本发明所要解决的技术问题是:现有技术中针对肺癌结节良恶性进行诊断过程中缺少无创方式的检测手段,导致了非必要的手术、增加了患者负担。
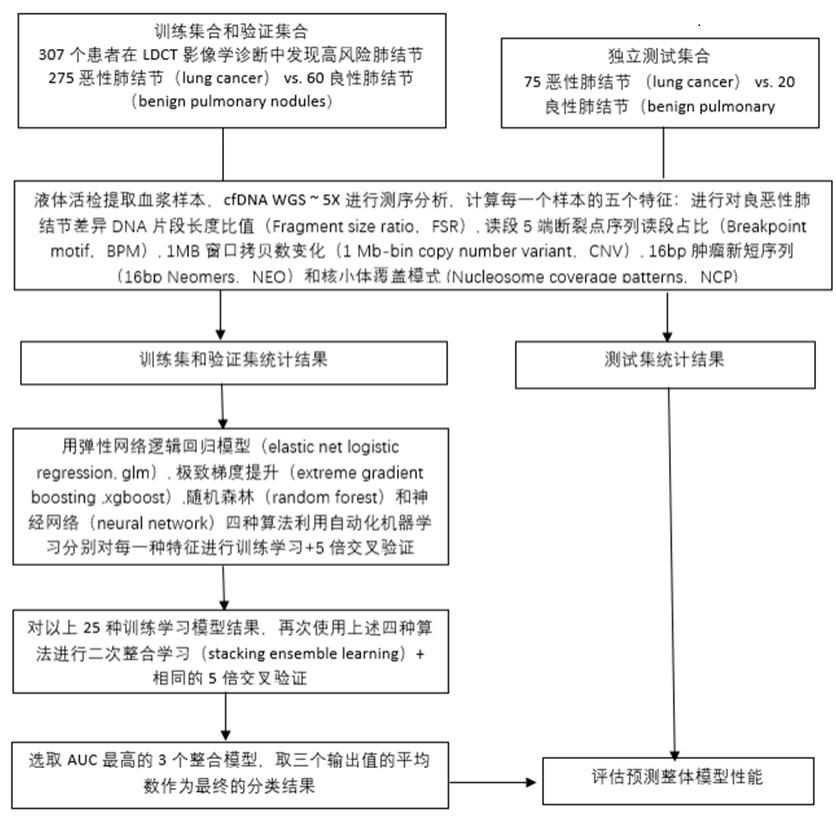
2、本专利的技术方案中,提供了一种对血浆样本cfdna进行wgs测序,通过对高通量测序结果,获取片段信息,进行对良恶性肺结节差异dna片段长度比值(fragment sizeratio), 读段5端断裂点序列读段占比(breakpoint motif), 1mb 窗口拷贝数变化(1 mb-bin copy number variant), 16bp肿瘤新短序列(16bp neomers)和核小体覆盖模式(nucleosome coverage patterns),利用弹性网络逻辑回归模型(elastic net logisticregression, glm), 极致梯度提升(extreme gradient boosting,xgboost),随机森林(random forest)和神经网络(neural network)四种算法利用自动化机器学习构建多特征多算法整合模型,实现对恶性肺结节无创精准诊断。
3、具体的技术方案是:
4、基因标志物在制备恶性肺结节筛查试剂中的应用;
5、所述的基因标志物包括:
6、第一标志物:cfdna片段比对至参考基因组的不同窗口中的短读段数量占比和长读段数量占比;
7、第二标志物:不同种类的cfdna片段比对至参考基因组的5’端的m个碱基片段在全部碱基片段中的占比;
8、第三标志物:wgs数据中染色体上不同窗口中的拷贝数;
9、第四标志物:肿瘤新短序列占比;
10、第五标志物:核小体覆盖模式。
11、所述的第一标志物通过如下步骤获得:将读段数据结果比对至参考基因组,将参考基因组划分为多个窗口,并分别获得在每个窗口范围内的短读段数量和超长读段数量占比。
12、所述的第二标志物通过如下步骤获得:将读段数据中的5’端的m个碱基数据作为碱基片段集合,并得到各种碱基片段在全部片段中所占比例。
13、所述的第三标志物通过如下步骤获得:将参考基因组划分为多个窗口,并分别获得wgs数据在1-22号染色体上不同窗口中的拷贝数数据。
14、所述的第四标志物通过如下步骤获得:
15、穷举法生成长度为16bp的短序列集合a;在人类参考基因序列中穷举出所有的长度为16bp短序列集合b,从集合a中将集合b数据剔除后,定义为无效子;
16、从癌症数据库中获得不同癌种的样本wgs测序结果,提取出多次出现的碱基替换突变; 根据碱基替换的位置,从无效子中找到包含这些碱基替换的无效子短序列集合c;
17、获取东亚人群中频率大于0.01的碱基替换突变;根据碱基替换的位置,从无效子中找到包含这些碱基替换的无效子短序列集合d;从集合c中将集合d的无效子序列排除,定义为新短序列;
18、统计出样本中能够读取到任意一个新短序列的样本数量,再针对每一个新短序列,搜索出包含这些新短序列的样本数量,将每一个新短序列的样本数量与所有能读取到任意新短序列的样本总数的比例。
19、所述的第五标志物通过如下步骤获得:
20、从gtrd数据库中获得转录因子,并从中排除掉不在cis-bp数据库中有已知转录位点的转录因子;
21、将获得的转录因子的转录位点附近-5kb到+5kb范围作为窗口,获得可以比对至这些窗口中的长度为100-220bp的片段,对窗口中的读段数据依次进行gc校正和测序深度平滑处理,得到每个转录因子的覆盖模式曲线;
22、对于每个转录因子,获得如下三个特征,共同作为核小体覆盖模式:
23、1)对于转录因子的全部转录位点,求出转录位点的上端1kb到下端1kb的平均深度;
24、2)对于获得的覆盖模式曲线,获得曲线波谷的幅度值,作为转录因子的中心深度;
25、3)对于获得的覆盖模式曲线进行快速傅里叶变换,获得核小体振幅信号的最高点的振幅数值。
26、恶性肺结节筛查模型的构建方法,包括如下步骤:
27、步骤1,对阳性组和对照组的样本进行cfdna的提取并测序,获得读段数据;
28、步骤2,将读段数据结果比对至参考基因组,将参考基因组划分为多个窗口,并分别获得在每个窗口范围内的短读段数量占比和长读段数量占比,作为第一特征集合;
29、步骤3,将读段数据中的5’端的m个碱基数据作为碱基片段集合,并得到各种碱基片段在全部片段中所占比例作为第二特征集合;
30、步骤4,将参考基因组划分为多个窗口,并分别获得在每个窗口范围内的拷贝数数据,作为第三特征集合;
31、步骤5,将读取到16bp新短序列的的样本数与所有能读取到任意新短序列的样本总数的比例,作为第四特征集合;
32、步骤6,所选取的转录因子的核小体覆盖模式特征,作为第五特征集合;
33、步骤7,以第一、第二、第三、第四和第五特征集合共同作为初始特征值,作为模型特征向量输入至分类模型中,并以肺结节良恶性作为输出值,对模型进行训练,获得早筛模型。
34、所述的步骤3中包括:
35、步骤3-1,将参考基因组划分为多个窗口,并分别获得每个窗口范围内的长读段数量和短读段数量;
36、步骤3-2,将步骤3-1的所有窗口的短读段数量和长读段数量标准化处理,标准化后的短读段数和长读段数的比例作为第一特征值。
37、所述的步骤3-1中窗口大小是5mb,共划分出541个窗口。
38、所述的短读段是指长度100-150bp,所述的长读段是151-220bp。
39、所述的步骤3中,m是4。
40、所述的步骤4中窗口大小是1mb,共划分出2475个窗口。
41、所述的步骤5 中,作为第四特征集合的获取步骤如下:
42、步骤5-1,穷举法生成长度为16bp的短序列集合a;在人类参考基因序列中穷举出所有的长度为16bp短序列集合b,从集合a中将集合b数据剔除后,定义为无效子;
43、步骤5-2,从癌症数据库中获得不同癌种的样本wgs测序结果,提取出多次出现的碱基替换突变; 根据碱基替换的位置,从无效子中找到包含这些碱基替换的无效子短序列集合c;
44、步骤5-3,获取东亚人群中频率大于0.01的碱基替换突变;根据碱基替换的位置,从无效子中找到包含这些碱基替换的无效子短序列集合d;从集合c中将集合d的无效子序列排除,定义为新短序列;
45、步骤5-4,统计出样本中能够读取到任意一个新短序列的样本数量,再针对每一个新短序列,搜索出包含这些新短序列的样本数量,将每一个新短序列的样本数量与所有能读取到任意新短序列的样本总数的比例,作为模型的第四特征集合。
46、所述的癌症数据库是pcawg数据库。
47、不同癌种是指肠癌、肺癌、乳腺癌、胃癌、前列腺癌和肝癌。
48、东亚人群中的碱基替换突变是通过gnomad数据库获得。
49、所述的步骤6包括:
50、步骤6-1,从gtrd数据库中获得转录因子,并从中排除掉不在cis-bp数据库中有已知转录位点的转录因子;
51、步骤6-2,将步骤6-1中获得的转录因子的转录位点附近-5kb到+5kb范围作为窗口,获得可以比对至这些窗口中的长度为100-220bp的片段,对窗口中的读段数据依次进行gc校正和测序深度平滑处理,得到每个转录因子的覆盖模式曲线;
52、步骤6-3,对于每个转录因子,获得如下三个特征,共同作为核小体覆盖模式特征:
53、1)对于转录因子的全部转录位点,求出这些转录位点的上端1kb到下端1kb的平均深度;
54、2)对于获得的覆盖模式曲线,获得曲线波谷的幅度值,作为转录因子的中心深度;
55、3)对于获得的覆盖模式曲线进行快速傅里叶变换,获得核小体振幅信号的最高点的振幅数值。
56、所述的步骤7中,分类模型的步骤包括:
57、步骤7-1,分别将第一、第二、第三、第四和第五特征集合输入至不同的分类器模型中,进行模型的训练,并获得分别针对第一、第二、第三、第四和第五特征集合的最优的一个或多个分类器模型;
58、步骤7-2,将步骤7-1中获得的第一、第二、第三、第四和第五特征集合的最优分类器模型进行二次集合训练,构建出集成分类器模型。
59、所述的不同的分类器模型选自弹性网络回归模型(elastic net logisticregression, glm)、极致梯度提升(extreme gradient boosting, xgboost)、随机森林(random forest)、深度学习神经网络模型(deeplearning,nn)。
60、二次集合训练中采用广义线性模型(generalized linear model,glm),极致梯度提升xgboost模型或者深度学习回归模型。
61、一种恶性肺结节检测装置,包括:
62、测序模块,用于对阳性组和对照组的样本进行cfdna的提取并测序,获得读段数据,并且获得wgs测序数据;
63、第一特征获取模块,用于将读段数据结果比对至参考基因组,将参考基因组划分为多个窗口,并分别获得在每个窗口范围内的短读段数量和超长读段数量占比,作为第一特征集合;
64、第二特征获取模块,用于将读段数据中的5’端的m个碱基数据作为碱基片段集合,并得到各种碱基片段在全部片段中所占比例作为第二特征集合;
65、第三特征获取模块,用于将参考基因组划分为多个窗口,并分别获得wgs数据在染色体上不同窗口中的拷贝数数据,作为第三特征集合;
66、第四特征获取模块,将读取到16bp新短序列的的样本数与所有能读取到任意新短序列的样本总数的比例,作为第四特征集合;
67、第五特征获取模块,分析所选取的转录因子的核小体覆盖模式特征,作为第五特征集合;
68、预测模块,用于以第一、第二、第三、第四和第五特征集合共同作为初始特征值,作为模型特征向量输入至分类模型中,并以肺结节良恶性作为输出值,对模型进行训练,获得早筛模型。
- 还没有人留言评论。精彩留言会获得点赞!