基于深度学习和计算模拟的蛋白质变构调节剂的识别方法
本发明涉及g蛋白偶联受体变构调节剂的识别,具体的说,是一种基于深度学习和计算模拟的蛋白质变构调节剂的识别方法。
背景技术:
1、变构机制提供了一种调节受体功能的新范例,因此变构调节剂的合理设计越来越受到关注。蛋白质的正构位点是内源性激活配体结合的位点。正构位点的强进化保守性导致正构配体的交叉反应性问题,这可能导致不良的治疗副作用。而变构调节剂结合到一个与正构位点在拓扑上不同的位点,因此不与正构配体竞争。变构位点的进化保守性低于正构位点,变构调节剂可能在亚型选择性和特异性方面表现出更优的特性,与正构配体相比可能会减少副作用。蛋白质的变构调节是相当精细的。例如,正变构调节剂可以通过四种不同的方式增强下游信号传导:(1)促进正构激动剂结合亲和力但不直接影响信号传导,(2)直接增强信号传导而不影响正构激动剂结合,(3)增加正构配体结合亲和力并同时增加自身的信号传导,以及(4)降低正构配体结合亲和力但自身增加信号传导。负变构调节剂可以使用类似的组合来减少下游信号。变构调节剂稳定了蛋白质集合的独特构象,为受体提供了新的药理学。因此,越来越多的变构调节剂被发现作为潜在的药物。
2、然而,由于发现过程非常具有挑战性,只有少数变构调节剂被批准作为药物或用于临床试验。这是因为从药理学实验中检测调节剂的变构行为是一个具有挑战性的过程,而且使用突变实验确定变构调节剂的结合位点经常观察到假阳性。自2013年以来,解析复合物结构一直是识别gpcr中变构调节剂结合位点和姿势的最成功方法,然而其耗费巨大。近年来,结构生物学和计算技术的发展已经揭示了大量靶点的变构机制,这使得合理设计变构调节剂成为药物发现的新途径。
3、变构位点的识别是基于结构的变构调节剂虚拟筛选的前提,然而变构位点通常是神秘的,在被解析的蛋白质结晶中难以被发现。未结合配体的apo结构中通常不存在变构口袋,并且只有在配体存在时,变构位点松弛状态才在构象集合中占主导地位。分子模拟是成为生成构象集合和探索这些位点的有效方法。将位点预测与基于md的gpcr构象集合结合起来可能检测到静态实验结构中不明显的位点,这对于发现新的变构位点很有吸引力。
4、尽管计算机辅助变构调节剂的设计已经有了一些应用,但是仍旧存在以下问题:
5、变构效应通常通过微秒(μs,10-6)或毫秒(ms,10-3)时间尺度内发生,经典的常规分子动力学模拟(cmd)一般只能捕捉纳秒(ns,10-9)时间尺度的构象变化,因此可能难以捕捉到隐秘的变构位点。
6、为了捕捉到隐秘的变构位点需要进行广泛的分子动力学模拟,这将产生大量的模拟数据。以人工方式分析这些数据是非常困难耗时且带有一定的先验知识偏差。筛选获取有价值的构象中间态是快速有效预测变构位点的先决条件。
7、虚拟筛选可以筛选出结合得最稳定的小分子,但是无法确定该小分子的属性及其变构调节机制。而变构调节剂的作用机制则是相关研究中最为关心的一个课题,并且由于其复杂性难以在实验中被揭示。
技术实现思路
1、本发明的目的在于提供一种基于深度学习和计算模拟的蛋白质变构调节剂的识别方法,识别蛋白质潜在的变构位点、筛选出潜在的变构调节剂,进而研究其变构调节机制。
2、本发明通过下述技术方案解决上述问题:
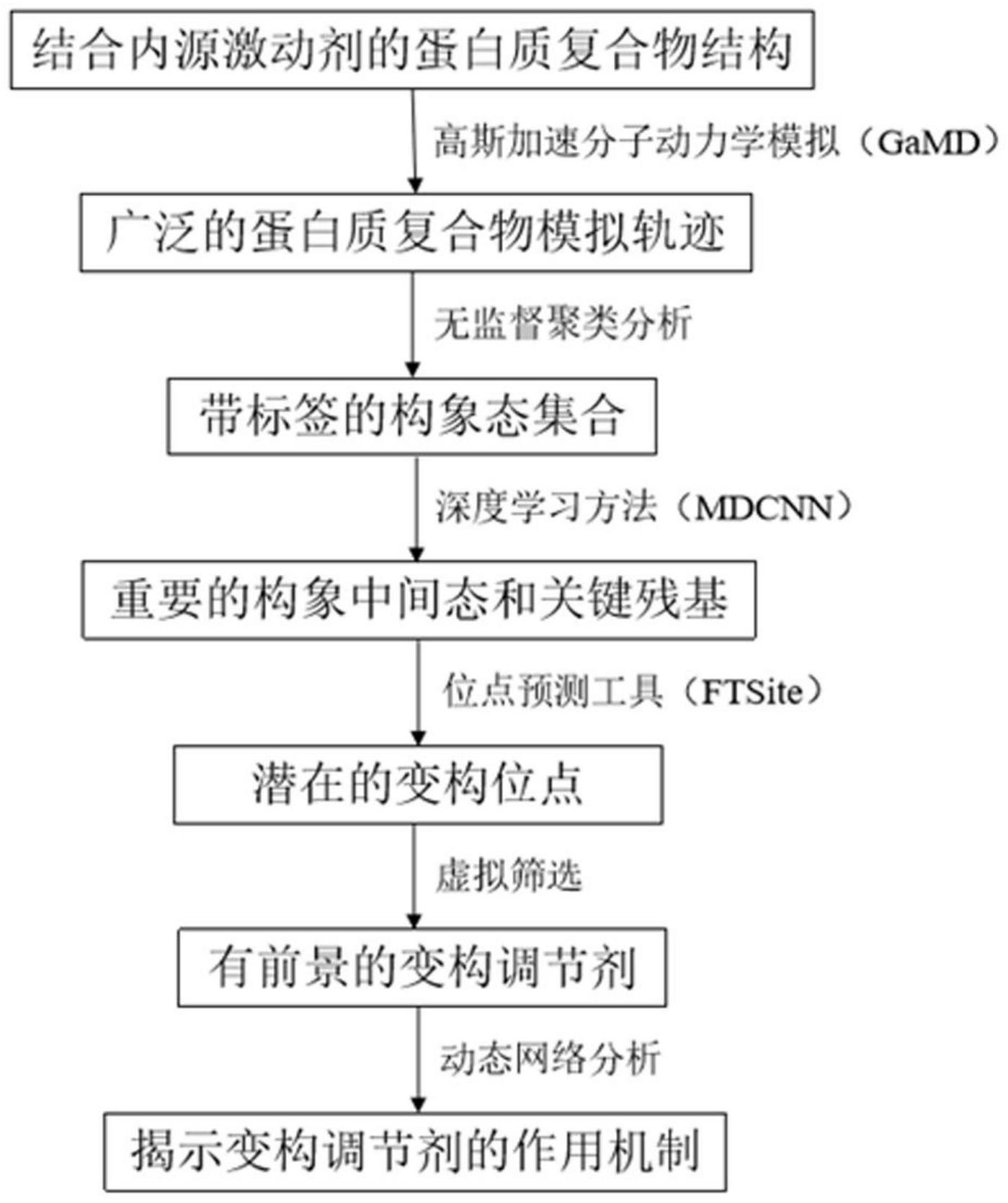
3、一种基于深度学习和计算模拟的蛋白质变构调节剂的识别方法,包括:
4、步骤s100、使用高斯加速分子动力学模拟获取结合内源性激动剂的蛋白质复合物的md模拟轨迹;高斯加速分子动力学模拟是一种增强采样方式,通过添加势能降低能垒的方式,可以在纳秒的模拟时间尺度内采样到毫秒级别的构象变化特征,因此可以捕捉到处于毫秒时间尺度上的变构位点;
5、步骤s200、使用无监督聚类分析将蛋白质复合物的md模拟轨迹进行初步分类,并且生成聚类标签;
6、步骤s300、将md模拟轨迹和聚类标签输入基于cnn的分类模型mdcnn中,从md模拟轨迹中识别出不同的构象态,mdcnn中的模型解释器lime在识别功能状态的同时寻找到每个构象态的关键结构和关键残基,借助lime反馈的关键残基辅助挑选有价值的构象态,用于后续变构位点预测;
7、步骤s400、将挑选出的构象态输入位点预测工具ftsite进行变构位点预测,除去正构位点之外得分最高的位点被认为是潜在的变构位点;
8、步骤s500、针对预测得到的潜在的变构位点使用基于结构的虚拟筛选得到结合最稳定的潜在药物分子;
9、步骤s600、借助动态网络分析揭示潜在药物分子的变构调控机制,确认潜在药物分子的性质,动态网络分析能够识别对结构信息传递起重要作用的变构路径和重要残基,从而揭示潜在药物分子的变构调节机制。
10、所述步骤s100具体包括:
11、步骤s110、获取目标蛋白质的非激活态晶体结构;
12、步骤s120、删除晶体结构中除了目标蛋白以外的其他组分,重新构建晶体结构中缺失的结构区域,使得目标蛋白质的结构完整;
13、步骤s130、获取蛋白质内源性激动剂的结构,然后使用分子对接并选取得分最高的合理对接位姿构建蛋白质-内源性激动剂的复合物结构;
14、步骤s140、在目标蛋白质的生理环境下对蛋白质和配体进行质子化,构建一个与生理环境类似的模拟体系,一般包括蛋白质复合物、溶剂分子、离子、(脂质膜成分);
15、步骤s150、针对构建完成的模拟体系,经过系统最小化和加热之后,在npt系综下进行无约束的cmd将模拟体系运行至一个相对平衡的状态。cmd平衡后的最后一个结构作为高斯加速分子动力学模拟的起始结构,开始运行高斯加速分子动力学模拟程序。
16、所述步骤s200具体包括:
17、步骤s210、从蛋白质复合物gamd轨迹中,间隔提取蛋白质的构象从而形成可以代表整段轨迹的蛋白质构象集。根据研究体系计算用于区分构象态的构象特征;
18、步骤s220、将构象特征作为聚类指标,使用无监督的聚类分析算法对蛋白质构象进行聚类,选出最佳的聚类结果后续作为蛋白质构象集的标签在mdcnn模型中使用。
19、所述步骤s300具体包括:
20、步骤s310、数据处理:使用s210中得到的蛋白质构象集,使用蛋白质cα原子叠加以消除整体旋转和平移。删除所有的氢原子,然后将其他原子的坐标被转成rgb坐标,从而得到数据集;
21、步骤s320、添加标签:将s220中得到的蛋白质构象集聚类结果作为数据集的标签数据读入,数据集与数据标签一一对应,以标明数据集中的构象属于哪一类;
22、步骤s330、数据集划分:对数据进行分组以消除模拟时序的影响,然后按照一定的比例随机划分为训练集、验证集。对数据集进行k折划分,得到k折交叉验证数据集;
23、步骤s340、模型的构建:将数据集作为输入,训练基于卷积神经网络(cnn)的用于蛋白质构象态分类识别的模型mdcnn,训练和验证过程采用k折交叉验证,然后使用准确度acc来评估分类器的性能;
24、步骤s350、构建模型解释器:构造lime解释器通过局部线性拟合的方式对mdcnn的预测结果进行解释;
25、步骤s360、将残基中包含的所有原子得分相加,得到蛋白质每个残基的重要性评分,通过排序选取评分排名前列的残基,视为该类构象态的重要残基。
26、所述步骤s400具体包括:
27、步骤s410、根据mdcnn反映的各构象态的重要残基,将其投影到蛋白质结构中,使用pymol进行可视化挑选出有价值的构象中间态,
28、步骤s420、从目标构象中间态轨迹中提取代表性的结构保存为pdb格式;
29、步骤s430、将代表性蛋白质结构文件上传至ftsite的服务器上(https://ftsite.bu.edu/),得到三个潜在的配体结合位点预测结果,除正构位点之外的得分最高的位点作为潜在的变构位点用于虚拟筛选。
30、所述步骤s500具体包括:
31、步骤s510、准备用于虚拟筛选的小分子数据集,生成对接盒子使其恰好完全覆盖潜在的变构位点中的全部残基,启动基于结构的虚拟筛选;
32、步骤s520、小分子的对接打分结果结合人工检查或精度更高的结合自由能计算等多种方式,选取与蛋白质结合最稳定的分子视为潜在的变构调节剂,输出蛋白质-变构调节剂复合物结构。
33、所述步骤s600具体包括:
34、步骤s610、对s500中得到的蛋白质-变构调节剂复合物在生理环境下构建模拟体系,使用分子动力学模拟获得一段仿真轨迹;
35、步骤s620、生成动态网络:将蛋白质-变构调节剂复合物的仿真轨迹输入vmd软件中,使用networkview插件生成一个动态网络;
36、步骤s630、利用动态网络分析潜在药物分子的变构调控机制,包括:
37、社区分析:在vmd中使用girvan-newman算法将动态网络进一步划分成不同的子网络,在vmd中对社区网络进行可视化分析,从而获取变构调节剂影响下,蛋白质各结构域的通信网络分布情况;
38、最短路径分析:使用vmd中的subopt程序,利用floyd-warshall算法搜索蛋白质网络中两个节点之间距离最短的路径。最短路径往往是最可能的或生物学相关的信号传递路径,由此可以得到变构调节剂作用下,蛋白质功能结构域之间的变构通讯路径,从而揭示变构调节剂的变构调控机制。
39、本发明与现有技术相比,具有以下优点及有益效果:
40、(1)本发明借助分子动力学模拟、深度学习、虚拟筛选和动态网络分析,识别蛋白质潜在的变构位点、筛选出潜在的变构调节剂和研究其变构调节机制。以β2型肾上腺素受体(β2ar)为例,我们成功识别了一个β2ar的新型变构位点,其位于高度保守的分子开关w6.48和d2.50之间。针对该位点筛选得到一个潜在的负变构调节剂zinc5042,它可以将受体稳定在非激活状态,与正构激动剂存在负协同效应。本发明同样适用于其他变构调节剂的研究。
41、(2)本发明使用高斯加速分子动力学模拟克服传统分子动力学模拟的时间尺度限制,可以采样到毫秒级别尺度上的构象变化,因此有利于捕捉潜在的变构位点。高斯加速分子动力学模拟无需手动设置加速参数,操作简便快捷。高斯加速分子动力学模拟能够通过添加势能降低能垒的方式,可以在纳秒的模拟时间尺度内采样到毫秒级别的构象变化,这大大降低了计算花费。
42、(3)本发明将无监督聚类和有监督的基于卷积神经网络的分类模型(mdcnn)相结合,实现了分子模拟轨迹的自动化处理。使用者只需将轨迹输入到模型中进行运算,而无需复杂的预处理,mdcnn可以自动化完成建模、解释、分析过程,大大提高模拟轨迹的分析效率,而且能够有效避免人工分析的偏差。同时mdcnn中整合了lime解释器对cnn模型进行解释,这能够帮助我们捕获不同构象态之间的特异性和重要残基分布。这有利于帮助我们筛选和识别出有价值的中间态以用于后续变构位点预测。
43、(4)本发明整合了动态网络分析用于揭示变构调节剂的作用机制,动态网络分析可以分析变构信息传递效率,识别对结构信息传递起重要作用的变构路径以及重要残基,从而揭示变构调节剂的药物属性和作用机制,辅助领域内的实验和理论研究。
- 还没有人留言评论。精彩留言会获得点赞!