一种基于深度学习的药物表征方法及存储介质
1.本发明涉及计算机辅助药物设计与深度学习技术领域,提供了一种基于深度学习的药物表征方法及存储介质。
背景技术:
2.在药物开发和再利用过程中,进行各项实验,如药物化学实验、动物实验和临床实验等,是非常耗时而且昂贵的过程。随着大量药物实验数据的产生和计算机技术的发展,使用基于数据和计算模型的方法进行计算机辅助药物设计是目前药物研发过程中的重要方法,其可以进行初步筛选,以减少实验次数,降低研发成本,缩短研发周期。在计算机辅助药物设计中最重要、最关键的技术就是药物表征。
3.近些年来,药物表征方法主要有分子描述符和自监督学习方法来获取表征,但仍存在适应性差、计算量大以及难以训练等局限性。
4.基于分子描述符的药物表征方法是目前最主流的方法,其主要是通过计算统计药物分子中各种子结构的类别和数量得到一个二进制的表征向量。此方法一般联合传统机器学习方法共同使用,具备一定的表征能力,但是不具备自适应性。此类药物表征方法,一旦选定分子描述符,则生成的药物表征向量固定,无法根据不同的药物筛选任务来学习最合适的表征向量,而且不同分子描述符会严重影响筛选任务的性能。
5.基于自监督学习的药物表征方法是主要分为两种,分别是基于分子序列模型和基于分子图模型的自监督表示学习方法。这类方法都是通过遮蔽复原策略来先预训练一个超大规模的模型,然后使用预训练后的全局特殊词元的向量作为药物表征向量。此方法使用的预训练模型参数量非常大(通常数亿甚至数百亿),因此需要巨大的数据和海量的计算资源才能训练,随之而来的还有收敛速度慢、训练时间长和药物表征生成时计算复杂度高等一系列问题,在人力、物力、时间上耗费巨大。
6.基于扩展连接指纹的有监督训练卷积神经网络模型,使用分子指纹作为监督信号,监督卷积神经网络模型的表示学习,可以根据不同的药物筛选任务,生成不同的药物表征。该方法使用了卷积神经网络模型和监督学习预训练策略,不仅计算量大大减少,且提升了药物表征的普适性,同时模型具有易训练和可解释性的特点。但该技术存在的缺点是需要计算生成监督标签,耗费时间较长。
技术实现要素:
7.针对上述研究的问题,本发明的目的在于提供一种基于深度学习的药物表征方法,解决现有技术中药物表征适应性差、不可学习,训练开销巨大的问题。
8.为了达到上述目的,本发明采用如下技术方案:
9.本发明提供了一种基于深度学习的药物表征方法,包括如下步骡:
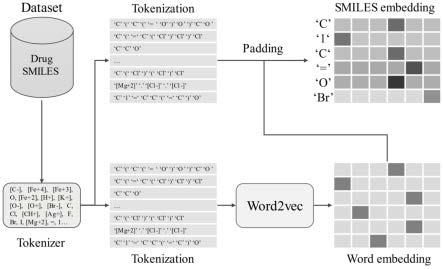
10.1)药物分子smiles数据集的收集;2)药物分子smiles数据增强;3)药物分子smiles分词;4)词元嵌入初始化;5)设计卷积神经网络模型;6)药物分子指纹特征生成;7)
预训练卷积神经网络模型;8)经过迭代计算,导出预训练模型;9)将分子输入表征模型得到静态分子表征,或共同训练动态表征以适应下游任务。
11.步骤1:获取chembl数据库中提供的药物分子规范smiles和药物编号构建数据集,其中随机选取80%药物作为训练集,10%做为验证集,10%作为测试集;
12.步骤2:使用rdkit工具包读取药物分子,并随机生成5个药物分子smiles,得到数据增强后的随机药物分子smiles;
13.步骤3:构建一个smiles分词器对药物分子smiles进行分词,得到词元化后的药物分子smiles列表;
14.步骤4:将步骤3得到的药物分子smiles列表进行onehot编码后使用word2vec模型得到词元的嵌入向量;
15.步骤5:构建基于卷积神经网络的深度学习模型,得到卷积神经网络模型;
16.步骤6:计算每一个药物分子的扩展连接分子指纹;
17.步骤7:使用扩展连接指纹作为标签监督训练卷积神经网络模型;
18.步骤8:保存卷积神经网络模型除输出层外的所有权重和偏置参数,导出预训练模型;
19.步骤9:将药物分子smiles输入预训练模型得到静态药物表征,或共同训练动态药物表征以适应下游任务。
20.上述技术方案中,所述步骤2的具体步骤为:
21.采用rdkit工具包将每一个药物分子的规范smiles读入成mol文件,用mol文件唯一表示一个药物分子;随机初始节点的原子,通过药物分子mol文件生成5个随机药物分子smiles,得到数据增强后的随机药物分子smiles。
22.上述技术方案中,所述步骤3的具体步骤为:
23.将步骤1和步骤2中收集到的药物分子smiles全部按照字符切分,得到切分后的字符列表;
24.将字符列表进行字节对编码,选择出现频次最高的一对相邻字符数据作为单位数据,即词元,并用该单位数据替换原有相邻字符,重复迭代多次,直到已有新单位数据达到词汇表大小v=2000,或达到设定迭代最大次数m=60000。;
25.保存已满足迭代停止条件的词汇表,并且联合词汇表和原子正则表达式token_regex,共同作为smiles分词器,其中原子正则表达式token_regex为:
26.token_regex=(\[[^\]]+]|br?|cl?|n|o|s|p|f|i|b|c|n|o|s|p|\(|\)|\.|=|#|-|\+|\\\/|:|~|@|\?|>|\*|\$|\%[0-9]{2}|[0-9])
[0027]
将步骤1和步骤2中收集到的药物分子smiles输入smiles分词器进行分词,得到词元化后的药物分子smiles列表。
[0028]
上述技术方案中,所述步骤4的具体步骤为:
[0029]
将已经词元化后的药物分子smiles列表进行onehot编码,得到相应的onehot向量;
[0030]
随机打乱所有药物分子smiles列表的onehot向量,将onehot向量分批次送入一个基于skipgram和负采样的word2vec模型进行训练,得到每个词元的嵌入向量。
[0031]
上述技术方案中,所述步骤5中卷积神经网络模型的基础结构采用textcnn模型,
具体设计为:
[0032]
textcnn模型接收词元化后的药物分子smiles列表对应的嵌入向量矩阵作为输入,词嵌入向量矩阵由步骤4中得到的嵌入向量按照药物分子smiles列表的词元顺序堆叠形成,设xi表示wordi的k维嵌入向量,则一条长为n的药物分子smiles列表的嵌入向量矩阵可以表示为:
[0033][0034]
其中,表示拼接(concat)操作,最终得到的x
1:n
为k
×
n的药物分子smiles列表的嵌入向量矩阵,在卷积操作中,卷积核通过一个长为h的窗口在药物分子smiles列表的嵌入向量矩阵上获取对应长度的特征:
[0035]ci
=f(w
·
x
i:i+h-1
+b)
[0036]
其中ci是卷积层提取的第i个特征,w是神经网络中的权重矩阵,是偏置项,f是一个非线性的激活函数,x
i:i+h-1
表示药物分子smiles列表的嵌入向量矩阵的第i行到第i+h行;
[0037]
在卷积计算过程中,这个卷积核会以固定的长度在药物分子smiles列表的嵌入向量矩阵中滑动计算得到对应的feature map,通过设置不同尺度的卷积核,提取词向量矩阵中不同长度词组信息;textcnn模型采用的池化函数是最大池化(1-max),最大池化就是在提取的特征向量中选取一个最大特征从而代替整个向量,然后将这些特征拼接起来构成一个新的特征向量;
[0038][0039]
其中表示最大特征,cj表示特征向量中的每一个值。
[0040]
上述技术方案中,所述步骤6的具体步骤为:
[0041]
对于每一个药物分子,使用rdkit工具包计算扩展连接分子指纹(extended connectivity fingerprints),设置扩展连接指纹的半径为3,指纹长度为1024位,得到1024位的二进制分子指纹向量。
[0042]
上述技术方案中,所述步骤7的具体步骡为:
[0043]
将步骤5的卷积神经网络模型输出向量长度设置为1024,即扩展连接指纹的长度;将卷积神经网络模型的最后输出作为分子特征向量,扩展连接指纹作为监督标签,通过cosine距离来计算两者的差异,计算公式为:
[0044]
loss=1-cosine(ecfp,o)
[0045]
其中loss表示扩展连接指纹和模型输出特征向量之间的差异,fcfp表示1024位的扩展连接分子指纹,o表示模型的输出;
[0046]
使用随机梯度下降法优化卷积神经网络模型参数,使扩展连接指纹和模型输出特征向量之间的差异逐步减小,直到其差异不再变化即模型训练完成。
[0047]
上述技术方案中,所述步骤9的具体步骤为:
[0048]
若需要直接得到药物表征,则将药物分子smiles输入预训练模型,即可得到相应的分子表征,此时分子表征向量长度为定长1024位;
[0049]
若需要将药物表征用于后续计算机辅助药物设计任务,则需要在已有分子表征模型后接一个前馈全连接神经网络进行特征变换,将1024位的药物表征向量变换到需要的维
度,再联合后续任务的标签进行联合学习训练。
[0050]
发明同现有技术相比,其有益效果表现在:
[0051]
本发明在预处理阶段采用了一种可学习的分词方式,包含了统计意义和药物化学语义,使药物分子smiles分词更加高效合理;在模型设计阶段采用了一个基于textcnn的模型,可训练的参数量小,同时又能够学习到普适性药物表征;使用了分子指纹作为监督信号的预训练方法,使计算量大大减小,具体为:
[0052]
1.本发明在预处理阶段使用了一种兼顾字节对编码和化学原子的分词方式,充分考虑了大规模药物分子smiles数据中标记字符的统计规律和化学中原子实体的语义信息,即分词后的词元既拥有较高的统计频率又包含有化学原子语义,同时能够得到长度更短、更易于计算的词元列表。
[0053]
2.本发明采用了卷积神经网络模型,模型设计基于textcnn模型家族,模型的卷积核长度分别设置为3,5,7,9,12,16,可以捕获多尺度的局部词元组合特征;与其他自监督深度学习模型相比,模型参数仅为200万,约为其他模型的1%;本发明采用的模型大大减少了参数量,提高了训练速度和药物表征向量生成速度。
[0054]
3.本发明采用了自生成有监督预训练方法,监督信号由药物分子计算生成,不需要人工打标签;有监督的预训练方法也仅需要在最后一步生成表征向量时计算损失值,不需要进行遮蔽复原训练,预训练预测次数减少了约98%;本发明采用的预训练方法大大减少了预训练预测次数、减少了训练时间,获得结果也更加直观合理。
附图说明
[0055]
图1数据预处理过程的操作流程图。
[0056]
图2为本发明中卷积神经网络的模型架构图。
[0057]
图3为本发明的技术流程图。
具体实施方式
[0058]
下面将结合附图及具体实施方式对本发明作进一步的描述。
[0059]
本发明实施提出了一种基于深度学习的药物表征方法,用于获得药物分子表征。
[0060]
本发明主要流程包括:1)药物分子smiles数据集的收集;2)药物分子smiles数据增强;3)药物分子smiles分词;4)词元嵌入初始化;5)设计卷积神经网络模型;6)药物分子指纹特征生成;7)预训练卷积神经网络模型;8)经过迭代计算,导出预训练模型;9)将分子输入表征模型得到静态分子表征,或共同训练动态表征以适应下游任务,具体实现步骤如下:
[0061]
一、药物分子数据集的收集
[0062]
从chembl数据库中提供的药物分子规范smiles和药物编号构建数据集,共有约180万药物分子,这部分数据集中随机选取80%药物作为训练集,训练集用于预训练阶段,10%作为验证集,10%作为测试集,验证集和测试集主要用于检验预训练模型的性能,以保证模型能够生成有效的药物表征向量。
[0063]
二、药物分子数据增强
[0064]
采用python语言的rdkit工具包将每一个药物分子的规范smiles读入成mol文件,
用mol文件唯一表示药物分子。
[0065]
以随机化初始原子的方式,通过药物分子mol文件生成5个随机smiles,得到数据增强后的随机smiles。
[0066]
三、药物分子smiles分词
[0067]
将步骤1和步骤2中收集到的药物分子smiles全部按照字符切分,得到切分后的字符列表;
[0068]
将字符列表进行字节对编码,选择出现频次最高的一对相邻字符数据作为单位数据,即词元,并用该单位数据替换原有相邻字符,重复迭代多次,直到已有新单位数据达到词汇表大小v=2000,或达到设定迭代最大次数m=60000。;
[0069]
保存已满足迭代停止条件的词汇表,并且联合词汇表和原子正则表达式token_regex,共同作为smiles分词器,其中原子正则表达式token_regex为:
[0070]
token_regex=(\[[^\]]+]|br?|cl?|n|o|s|p|f|i|b|c|n|o|s|p|\(|\)|\.|=|#|-|\+|\\\/|:|~|@|\?|>|\*|\$|\%[0-9]{2}|[0-9])
[0071]
将步骤1和步骤2中收集到的药物分子smiles输入smiles分词器进行分词,得到词元化后的药物分子smiles列表。
[0072]
四、词元嵌入初始化
[0073]
将已经词元化后的药物分子smiles列表进行onehot编码,得到相应的onehot向量;
[0074]
随机打乱所有药物分子smiles列表的onehot向量,将onehot向量分批次送入一个基于skipgram和负采样的word2vec模型进行训练,得到每个词元的嵌入向量。
[0075]
五、设计卷积神经网络
[0076]
textcnn模型接收词元化后的药物分子smiles列表对应的嵌入向量矩阵作为输入,词嵌入向量矩阵由步骤4中得到的嵌入向量按照药物分子smiles列表的词元顺序堆叠形成,设xi表示wordi的k维嵌入向量,则一条长为n的药物分子smiles列表的嵌入向量矩阵可以表示为:
[0077][0078]
其中,表示拼接(concat)操作,最终得到的x
1:n
为k
×
n的药物分子smiles列表的嵌入向量矩阵,在卷积操作中,卷积核通过一个长为h的窗口在药物分子smiles列表的嵌入向量矩阵上获取对应长度的特征:
[0079]ci
=f(w
·
x
i:i+h-1
+b)
[0080]
其中ci是卷积层提取的第i个特征,w是神经网络中的权重矩阵,是偏置项,f是一个非线性的激活函数,x
i:i+h-1
表示药物分子smiles列表的嵌入向量矩阵的第i行到第i+h行;
[0081]
在卷积计算过程中,这个卷积核会以固定的长度在药物分子smiles列表的嵌入向量矩阵中滑动计算得到对应的feature map,通过设置不同尺度的卷积核,提取词向量矩阵中不同长度词组信息;textcnn模型采用的池化函数是最大池化(1-max),最大池化就是在提取的特征向量中选取一个最大特征从而代替整个向量,然后将这些特征拼接起来构成一个新的特征向量;
[0082][0083]
其中表示最大特征,cj表示特征向量中的每一个值。
[0084]
六、药物分子指纹特征生成
[0085]
对于每一个药物分子,使用rdkit工具包计算扩展连接分子指纹(extended connectivity fingerprints),设置扩展连接指纹的半径为3,指纹长度为1024位,得到1024位的二进制分子指纹向量作为后续模型训练的监督标签信号。
[0086]
七、预训练卷积神经网络
[0087]
将步骤5的卷积神经网络模型输出向量长度设置为1024,即扩展连接指纹的长度;将卷积神经网络模型的最后输出作为分子特征向量,扩展连接指纹作为监督标签,通过cosine距离来计算两者的差异,计算公式为:
[0088]
loss=1-cosine(ecfp,o)
[0089]
其中loss表示扩展连接指纹和模型输出特征向量之间的差异,ecfp表示1024位的扩展连接分子指纹,o表示模型的输出;
[0090]
使用随机梯度下降法优化卷积神经网络模型参数,使扩展连接指纹和模型输出特征向量之间的差异逐步减小,直到其差异不再变化即模型训练完成。
[0091]
八、经过迭代计算,导出预训练模型
[0092]
在模型的损失几乎不再变化之后,模型即训练完成,使用随机梯度下降法优化模型参数,使扩展连接指纹和模型输出特征向量之间的差异尽可能的小,直到其差异不再变化即模型训练完成。
[0093]
使用深度学习框架的savemodel函数保存当前模型的所有可学习参数的字典并存为pth文件,在后续使用时导入模型对应参数的权重和偏置即可。
[0094]
九、将分子输入表征模型得到静态分子表征,或共同训练动态表征以适应下游任务
[0095]
若需要直接得到药物表征,则将药物分子smiles输入预训练模型,即可得到相应的分子表征,此时分子表征向量长度为定长1024位;
[0096]
若需要将药物表征用于后续计算机辅助药物设计任务,则需要在已有分子表征模型后接一个前馈全连接神经网络进行特征变换,将1024位的药物表征向量变换到需要的维度,再联合后续任务的标签进行联合学习训练。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1