临床检测样本的质控方法、装置、电子设备及存储介质与流程
1.本技术涉及高通量测序技术领域,特别涉及一种临床检测样本的质控方法、装置、电子设备及存储介质。
背景技术:
2.通过对血液样本进行rna测序可以辅助分析人体生理和健康状况,但是在实际应用中临床检测样本的质量受到多种因素的影响,当出现临床检测样本被污染、或提取临床检测样本的操作流程不规范的情况时,临床检测样本的质量较差。
3.因此,如何对临床检测样本的质量进行准确检测,提高数据的有效性是本领域技术人员目前需要解决的技术问题。
技术实现要素:
4.本技术的目的是提供一种临床检测样本的质控方法、一种临床检测样本的质控装置、一种电子设备及一种存储介质,能够对临床检测样本的质量进行准确检测,提高数据的有效性。
5.为解决上述技术问题,本技术提供一种临床检测样本的质控方法,该临床检测样本的质控方法包括:
6.对临床检测样本进行rna测序获得原始fastq数据文件,对所述原始fastq数据文件进行质控过滤得到目标fastq数据文件;
7.将所述目标fastq数据文件与参考基因组序列文件进行比对,得到bam文件;
8.根据所述bam文件确定比对率、rrna(核糖体rna)含量和globin rna(珠蛋白rna)含量;
9.利用所述bam文件进行测序深度检测,得到基因3’端测序深度和基因5’端测序深度;
10.对所述临床检测样本进行多种测序策略得到的多种测序数据进行一致性检验,得到一致性检验结果;
11.根据所述比对率、所述rrna含量、所述globin rna含量、所述基因3’端测序深度、所述基因5’端测序深度、以及所述一致性检验结果生成所述临床检测样本的质控结果。
12.可选的,对所述原始fastq数据文件进行质控过滤得到目标fastq数据文件,包括:
13.去除所述原始fastq数据文件中的目标接头序列和低质量序列,得到所述目标fastq数据文件;
14.其中,所述低质量序列包括质量值低于质量值阈值的序列和长度小于长度阈值的序列。
15.可选的,将所述目标fastq数据文件与参考基因组序列文件进行比对,得到bam文件,包括:
16.获取fasta格式的所述参考基因组序列文件,创建所述参考基因组序列文件的索
引序列,将所述目标fastq数据文件与所述索引序列进行比对,得到所述bam文件。
17.可选的,根据所述bam文件确定比对率,包括:
18.根据所述bam文件确定比对到所述参考基因组序列文件的目标序列;
19.将所述目标序列在所述目标fastq数据文件中所有序列的比例设置为所述比对率。
20.可选的,在根据所述bam文件确定比对率之后,还包括:
21.若所述比对率小于第一阈值,则生成判定所述临床检测样本不合格的质控结果。
22.可选的,在根据所述bam文件确定rrna含量和globin rna含量之前,还包括:
23.确定所述参考基因组序列文件中的rrna序列坐标和globin rna序列坐标;
24.按照所述rrna序列坐标和所述globin rna序列坐标统计n个对照血液样本中的对照rrna含量和对照globin rna含量;
25.根据n个所述对照血液样本的对照rrna含量确定第一标准含量区间;
26.根据n个所述对照血液样本的对照globin rna含量确定第二标准含量区间;
27.相应的,在根据所述bam文件确定rrna含量和globin rna含量之后,还包括:
28.若所述rrna含量不在所述第一标准含量区间内,则生成判定所述临床检测样本不合格的质控结果;
29.若所述globin rna含量不在所述第二标准含量区间内,则生成判定所述临床检测样本不合格的质控结果。
30.可选的,在利用对照血液样本和基因组注释文件对所述bam文件进行测序深度比对之前,还包括:
31.利用基因组注释文件获取基因3’端位置信息和基因5’端位置信息;
32.根据所述基因3’端位置信息确定m个对照血液样本中的基因3’端平均测序深度,并根据所述基因5’端位置信息确定m个所述对照血液样本中的基因5’端平均测序深度;
33.将同一基因对应的基因3’端平均测序深度与基因5’端平均测序深度的比值设置为特征值;
34.利用kmeans算法对所述特征值进行建模得到聚类模型,并根据肘部法确定所述聚类模型的聚类个数;
35.根据所述聚类模型确定距离阈值;
36.相应的,利用所述bam文件进行测序深度检测,得到基因3’端测序深度和基因5’端测序深度之后,还包括:
37.将所述基因3’端测序深度和所述基因5’端测序深度的比值设置为样本特征值;
38.计算所述样本特征值与所述聚类模型的聚类中心的欧氏距离;
39.若所述欧式距离大于所述距离阈值,则生成判定所述临床检测样本不合格的质控结果。
40.可选的,根据所述比对率、所述rrna含量、所述globin rna含量、所述基因3’端测序深度、基因5’端测序深度、以及所述一致性检验结果生成所述临床检测样本的质控结果,包括:
41.计算所述比对率对应的第一得分;
42.计算所述rrna含量对应的第二得分;
43.计算所述globin rna含量对应的第三得分;
44.计算所述基因3’端测序深度和基因5’端测序深度对应的第四得分;
45.计算所述一致性检验结果对应的第五得分;
46.将所述第一得分、所述第二得分、所述第三得分、所述第四得分和所述第五得分的总和设置为样本总得分,根据所述样本总得分生成所述临床检测样本的质控结果。
47.本技术还提供了一种临床检测样本的质控装置,该装置包括:
48.原始数据质控模块,用于对临床检测样本进行rna测序获得原始fastq数据文件,对所述原始fastq数据文件进行质控过滤得到目标fastq数据文件;
49.映射文件构建模块,用于将所述目标fastq数据文件与参考基因组序列文件进行比对,得到bam文件;
50.比对率检测模块,用于根据所述bam文件确定比对率;
51.目标rna含量统计模块,用于根据所述bam文件确定rrna含量和globin rna含量;
52.测序深度统计模块,用于利用所述bam文件进行测序深度检测,得到基因3’端测序深度和基因5’端测序深度;
53.一致性检验模块,用于对所述临床检测样本进行多种测序策略得到的多种测序数据进行一致性检验,得到一致性检验结果;
54.判断模块,用于根据所述比对率、所述rrna含量、所述globin rna含量、所述基因3’端测序深度、所述基因5’端测序深度、以及所述一致性检验结果生成所述临床检测样本的质控结果。
55.本技术还提供了一种存储介质,其上存储有计算机程序,所述计算机程序执行时实现上述临床检测样本的质控方法执行的步骤。
56.本技术还提供了一种电子设备,包括存储器和处理器,所述存储器中存储有计算机程序,所述处理器调用所述存储器中的计算机程序时实现上述临床检测样本的质控方法执行的步骤。
57.本技术提供了一种临床检测样本的质控方法,包括:对临床检测样本进行rna测序获得原始fastq数据文件,对所述原始fastq数据文件进行质控过滤得到目标fastq数据文件;将所述目标fastq数据文件与参考基因组序列文件进行比对,得到bam文件;根据所述bam文件确定比对率、rrna含量和globin rna含量;利用所述bam文件进行测序深度检测,得到基因3’端测序深度和基因5’端测序深度;对所述临床检测样本进行多种测序策略得到的多种测序数据进行一致性检验,得到一致性检验结果;根据所述比对率、所述rrna含量、所述globin rna含量、所述基因3’端测序深度、所述基因5’端测序深度、以及所述一致性检验结果生成所述临床检测样本的质控结果。
58.本技术对临床检测样本对应的原始fastq数据文件进行质控过滤并与参考基因组序列文件进行比对,得到bam文件,根据上述bam文件可以确定比对率、rrna含量和globin rna含量。本技术还利用对照血液样本和基因组注释文件对上述bam文件进行测序深度比对,得到临床检测样本的基因3’端测序深度和基因5’端测序深度。本技术还对所述临床检测样本进行多种测序策略得到的多种测序数据进行一致性检验,得到一致性检验结果。本技术将上述比对率、rrna含量、globin rna含量、基因3’端测序深度、基因5’端测序深度、以及所述一致性检验结果作为统计指标,综合判断临床检测样本是否合格。上述过程从多个
方面对临床检测样本的质量进行检测,结合多个统计指标综合判断临床检测样本是否合格,因此本技术能够对临床检测样本的质量进行准确检测,提高数据的有效性。本技术同时还提供了一种临床检测样本的质控装置、一种存储介质和一种电子设备,具有上述有益效果,在此不再赘述。
附图说明
59.为了更清楚地说明本技术实施例,下面将对实施例中所需要使用的附图做简单的介绍,显而易见地,下面描述中的附图仅仅是本技术的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
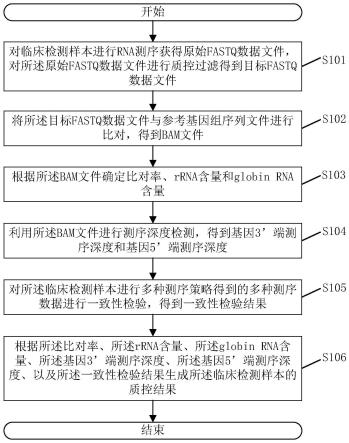
60.图1为本技术实施例所提供的一种临床检测样本的质控方法的流程图;
61.图2为本技术实施例所提供的一种rrna含量统计的箱形图;
62.图3为本技术实施例所提供的一种globin rna含量统计的箱形图;
63.图4为本技术实施例所提供的一种肘部法确定最佳聚类个数的示意图;
64.图5为本技术实施例所提供的一种异常样本可视化示意图;
65.图6为本技术实施例所提供的一种针对血液样本rnaseq质控的流程图;
66.图7为本技术实施例所提供的一种临床检测样本的质控装置的结构示意图。
具体实施方式
67.为使本技术实施例的目的、技术方案和优点更加清楚,下面将结合本技术实施例中的附图,对本技术实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本技术一部分实施例,而不是全部的实施例。基于本技术中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本技术保护的范围。
68.下面请参见图1,图1为本技术实施例所提供的一种临床检测样本的质控方法的流程图。
69.具体步骤可以包括:
70.s101:对临床检测样本进行rna测序获得原始fastq数据文件,对所述原始fastq数据文件进行质控过滤得到目标fastq数据文件;
71.其中,本实施例可以应用于能够实现基因处理任务的服务器,上述临床检测样本指用于进行rna测序的血液样本,在本步骤之前可以存在利用上述临床检测样本按照多种测序策略进行rna测序的操作。
72.本步骤获取的临床检测样本对应的原始fastq数据文件为fastq格式储存的序列信息。由于上述原始fastq数据文件中存在部分低质量的序列和接头序列,本步骤通过对原始fastq数据文件进行质控过滤得到目标fastq数据文件。
73.s102:将所述目标fastq数据文件与参考基因组序列文件进行比对,得到bam文件;
74.其中,在本步骤之前从公共基因数据库中下载fasta格式的参考基因组序列文件,将目标fastq数据文件与参考基因组序列文件进行比对,得到bam文件,即映射文件。
75.s103:根据所述bam文件确定比对率、rrna含量和globin rna含量;
76.其中,在得到bam文件的基础上,本步骤可以确定比对率、rrna含量和globin rna含量。上述比对率用于描述目标fastq数据文件比对到参考基因组序列文件的序列占总序
列的百分比。
77.s104:利用所述bam文件进行测序深度检测,得到基因3’端测序深度和基因5’端测序深度;
78.其中,参考基因组中存在多个基因,利用bam文件进行测序深度检测,可以得到每一基因的基因3’端测序深度和基因5’端测序深度。
79.s105:对所述临床检测样本进行多种测序策略得到的多种测序数据进行一致性检验,得到一致性检验结果;
80.其中,在本步骤之前可以存在利用上述临床检测样本按照多种测序策略进行测序操作,本实施例可以获取每一种测序策略对应的测序数据,并将所有的测序数据进行一致性检验。若所有测序策略得到的测序数据的一致性大于预设值(如95%),则判断通过一致性检验;反之则不通过一致性检验。
81.s106:根据所述比对率、所述rrna含量、所述globin rna含量、所述基因3’端测序深度、所述基因5’端测序深度、以及所述一致性检验结果生成所述临床检测样本的质控结果。
82.其中,本实施例可以将比对率、rrna含量、globin rna含量、基因3’端测序深度、基因5’端测序深度以及一致性检验结果作为统计指标,结合所有的统计指标进行综合判断,本实施例还可以将上述统计指标上传至人机界面,以便由用户进行决策,并根据上述决策输出临床检测样本是否合格的质控结果。上述质控结果可以为临床检测样本合格,也可以为临床检测样本不合格。作为一种可行的实施方式,若临床检测样本不合格,则可以判定样本被污染或样本提取操作不规范。
83.本实施例对临床检测样本对应的原始fastq数据文件进行质控过滤并与参考基因组序列文件进行比对,得到bam文件,根据上述bam文件可以确定比对率、rrna含量和globin rna含量。本实施例还利用对照血液样本和基因组注释文件对上述bam文件进行测序深度比对,得到临床检测样本的基因3’端测序深度和基因5’端测序深度。本实施例还对所述临床检测样本进行多种测序策略得到的多种测序数据进行一致性检验,得到一致性检验结果。本实施例将上述比对率、rrna含量、globin rna含量、基因3’端测序深度、基因5’端测序深度、以及所述一致性检验结果作为统计指标,综合判断临床检测样本是否合格。上述过程从多个方面对临床检测样本的质量进行检测,结合多个统计指标综合判断临床检测样本是否合格,因此本实施例能够对临床检测样本的质量进行准确检测,提高数据的有效性。
84.作为对于图1对应实施例的进一步介绍,本实施例可以通过以下方式对原始fastq数据文件进行质控过滤:去除所述原始fastq数据文件中的目标接头序列和低质量序列,得到所述目标fastq数据文件;其中,所述低质量序列包括质量值低于质量值阈值的序列和长度小于长度阈值的序列。
85.原始fastq数据文件中有些数据的质量较低,这些低质量的数据会影响后续数据分析准确性,因此需要去除。同时原始fastq数据文件中存在接头序列,这些序列不是目标序列,因此也需要去除。
86.对原始fastq数据文件的质控操作具体包括以下步骤a1和步骤a2的过程:
87.步骤a1:获取原始fastq数据文件。
88.原始fastq数据文件为fastq格式储存的序列信息,每1条reads的信息,可以分成4
行;下面是一个illumina平台测序的真实数据,其中包含了1条reads的信息:
89.第1行:@st-e00126:128:hjflhccxx:2:1101:7405:1133;
90.第2行:ttgcaaaaaatttctctcattntgtaggttgcctgttca ctctgatgatagtttgtnttgg;
91.第3行:+
92.第4行:ffkkkfkkfkf《kk《f,afkkkkk7ffk77《fkk,《f7k,,7af《ff7fkk7aa,7《fa;
93.第1行主要储存序列测序时的坐标等信息。第2行是测序得到的序列信息,一般用atcgn来表示,其中n表示荧光信号干扰无法判断到底是哪个碱基。第3行以“+”开始,可以储存一些附加信息,一般是空的。第4行储存的是质量信息,与第2行的碱基序列是一一对应的,其中的每一个符号对应的ascii值成为phred值,可以理解为对应位置碱基的质量值,越大说明测序的质量越好。
94.关于phred值的说明如下:测序仪进行测序的时候,会自动根据荧光信号的强弱给出一个参考的测序错误概率(error probility,p),本实施例将p取log10之后再乘以-10,得到的结果为q。比如,p=1%,那么对应的q=-10*log10(0.01)=20;把这个q加上33或者64转成一个新的数值,称为phred,最后把phred对应的ascii字符对应到这个碱基。如q=20,phred=20+33=53,对应的符号是“5”。本实施例将测序错误概率来表示测序的质量,每一个一位符号的ascii值表示每一位碱基的质量。
95.步骤a2:利用软件cutadapt根据提供的接头序列去除原始fastq数据文件的序列中的接头序列,并根据设定的质量值阈值去掉低质量的序列。
96.命令行如下:
97.cutadapt-a adapter1-a adapter2-e 0.1-o 1-m 36
‑‑
max-n 5-q 30,30-j nt-osample.trim.r1.fastq.gz-p sample.trim.r2.fastq.gz r1_raw r2_raw;
98.上述命令行中的参数说明如下:
[0099]-a:接头1序列;
[0100]-a:接头2序列;
[0101]-e:接头序列匹配允许的最大错配率;
[0102]-m:根据最短长度筛选哪些序列进行保留;
[0103]-o:adapter与reads最小overlap,才算成功识别;
[0104]
‑‑
max-n:一条序列中n的数量,如果大于该数值该序列会被丢弃;
[0105]-q:修剪低质量碱基的阈值,如果只有一个值,只修剪3’端,如果有两个值用逗号隔开,5’端和3’端都修剪。
[0106]-o:第一条输出文件;
[0107]-p:第二条reads的输出结果;
[0108]
r1_raw:第一条reads的输入文件;
[0109]
r2_raw:第二条reads的输入文件;
[0110]
上述过程的输入文件为原始fastq数据文件;使用的软件为cutadapt序列质控软件;输出的文件为质控后的文件(即,目标fastq数据文件)。
[0111]
一条序列的两端分别称为5’端和3’端,默认情况会对两端低质量的碱基进行修剪,某些情况下只修剪其中的一段,这个-q参数如果只提供一个阈值,就只会对3’端修剪,如果提供两个阈值,就会对两端的序列修剪。修剪3’端和5’端的作用就是把低质量的序列
去除,得到高质量的序列。
[0112]
作为对于图1对应实施例的进一步介绍,可以按照以下方式构建bam文件:获取fasta格式的所述参考基因组序列文件,创建所述参考基因组序列文件的索引序列,将所述目标fastq数据文件与所述索引序列进行比对,得到所述bam文件(即映射文件)。
[0113]
具体的,上述过程可以利用参考基因组序列文件和目标fastq数据文件进行全基因组比对生成bam文件,进而可以通过bam文件获取比对率,统计rrna含量、globin rna含量。
[0114]
上述bam文件的构建过程包括以下步骤b1至步骤b3的过程:
[0115]
步骤b1:从公共基因数据库(ncbi,ucsc,ensembl等)中,下载fasta格式的参考基因组序列文件(下文以ref.fa表示参考基因组序列文件);
[0116]
步骤b2:输入参考基因组序列文件ref.fa,采用序列比对软件创建参考基因组的比对索引,生成的相关文件包括ref.fa.fai,ref.fa.amb,ref.fa..ann,ref.fa.bwt,ref.fa.pac,ref.fa.sa;
[0117]
步骤b3:通过比对工具利用临床样本的fastq数据和和步骤b得到的索引序列进行参考基因组进行序列比对,得到样本的原始比对bam文件。
[0118]
上述过程的输入文件包括参考基因组序列文件和目标fastq数据文件使用的线管软件包括序列索引构建和比对软件,输出文件包括参考基因组序列文件与索引文件、以及bam文件。
[0119]
作为对于图1对应实施例的进一步介绍,可以通过以下方式确定比对率:根据所述bam文件确定比对到所述参考基因组序列文件的目标序列;将所述目标序列在所述目标fastq数据文件中所有序列的比例设置为所述比对率。在根据所述bam文件确定比对率之后,若所述比对率小于第一阈值,则可以生成判定所述临床检测样本不合格的质控结果。
[0120]
本实施例可以基于比对率的进行异常样本检测,例如可以通过样本的比对率来判断一个样本是否在建库测序中存在污染。具体的,本实施例可以获取检测样本的bam文件,利用公开软件计算比对到人类参考基因组的序列占总序列的百分比。根据经验判断如果该样本的比对于小于90%,认为该样本存在污染需要重新建库测序。
[0121]
作为对于图1对应实施例的进一步介绍,在根据所述bam文件确定rrna含量和globin rna含量之前,还执行以下操作:确定所述参考基因组序列文件中的rrna序列坐标和globin rna序列坐标;按照所述rrna序列坐标和所述globin rna序列坐标统计n个对照血液样本中的对照rrna含量和对照globin rna含量;根据n个所述对照血液样本的对照rrna含量确定第一标准含量区间;根据n个所述对照血液样本的对照globin rna含量确定第二标准含量区间;
[0122]
相应的,在根据所述bam文件确定rrna含量和globin rna含量之后,若所述rrna含量不在所述第一标准含量区间内,则生成判定所述临床检测样本不合格的质控结果;若所述globin rna含量不在所述第二标准含量区间内,则生成判定所述临床检测样本不合格的质控结果。
[0123]
对于血液样本rna测序数据,可通过实验手动在建库过程中去除rrna和globin rna,可以根据rrna、globin rna含量来判断rrna和globin rna消除是符合格。基于rrna、globin rna含量的异常样本检测包括以下步骤c1至步骤c4的过程:
[0124]
步骤c1:从公共基因数据库(ncbi,ucsc,ensembl等)中,下载参考基因组序列fasta格式文件(下文以ref.fa表示参考基因组序列文件);
[0125]
步骤c2:根据下载的人类参考基因组文件获取对应的rrna、globin rna序列,并获取这些序列的坐标生成位置信息文件。位置信息格式:染色体编号:基因起始坐标-基因终止坐标;
[0126]
步骤c3:获取rrna&globin rna含量的参考置信区间:根据步骤c2获取的位置信息文件利用公开软件统计本地自有80例血液rnaseq对照数据中rrna、globin基因表达占比,建立本地rrna&globin rna表达数据库。获取rrna&globin rna在本地对照中的平均值和置信区间[mean(平均值)
±3×
std(标准差)]。定义rrna含量占比大于0.133或globinrna含量占比大于0.132为rrna&globin rna实验消除异常样本;
[0127]
步骤c4:统计临床诊断检测样本rrna&globin rna含量,判断检测样本是否符合实验去除要求。
[0128]
请参见图2和图3,图2为本技术实施例所提供的一种rrna含量统计的箱形图,图3为本技术实施例所提供的一种globin rna含量统计的箱形图,图2和图3的纵坐标表示样本名称,横坐标rrna_freq表示rrna含量,横坐标globin_freq表示globin rna含量。
[0129]
上述实施例的输入文件包括bam文件、rrna和globin rna的位置信息文件,使用到的软件为根据位置信息提取映射文件的reads软件,输出的信息包括rrna和globin rna含量统计结果。
[0130]
作为一种可行的实施方式,在利用对照血液样本和基因组注释文件对所述bam文件进行测序深度比对之前,还执行以下操作:利用基因组注释文件获取基因3’端位置信息和基因5’端位置信息;根据所述基因3’端位置信息确定m个对照血液样本中的基因3’端平均测序深度,并根据所述基因5’端位置信息确定m个所述对照血液样本中的基因5’端平均测序深度;将同一基因对应的基因3’端平均测序深度与基因5’端平均测序深度的比值设置为特征值;利用kmeans算法对所述特征值进行建模得到聚类模型,并根据肘部法确定所述聚类模型的聚类个数;根据所述聚类模型确定距离阈值;
[0131]
相应的,利用所述bam文件进行测序深度检测,得到基因3’端测序深度和基因5’端测序深度之后,还可以将所述基因3’端测序深度和所述基因5’端测序深度的比值设置为样本特征值;计算所述样本特征值与所述聚类模型的聚类中心的欧氏距离;若所述欧式距离大于所述距离阈值,则生成判定所述临床检测样本不合格的质控结果。
[0132]
具体的,通过疾病关联基因3’端和5’端外显子上测序深度差异分析与实验rna完整性一致性分析,判断可能的建库过程导致的不均一性,尽可能消减实验造成的诊断的假阴性,因此基于基因3’端和5’端测序深度差异分析的异常样本检测包括以下步骤d1至步骤d7的过程:
[0133]
步骤d1:从公开数据库下载基因组注释文件,收集和疾病相关的基因文件,并获取每个基因第一个外显子前50bp作为5’端位置信息,最后一个外显子后50bp作为3’端位置信息;
[0134]
步骤d2:利用本地80例对照血液样本的bam文件和步骤d1获取的位置信息文件,统计每个样本不同基因3’端和5’端的平均测序深度,并把3’端和5’端的平均测序深度的比值作为度量差异的标准,比值越大表示3’端和5’端的测序深度差异越大,总共获取4658个特
征值;
[0135]
步骤d3:特征选择和模型训练。对4658特征值进行过滤,过滤掉3’端和5’端测序深度都很低的特征,共获取1933特征,利用python的kmeans算法模块对数据进行建模。
[0136]
步骤d4:根据肘部法(elbow plot)选择最佳聚类个数。kmeans是以最小化样本与质点平方误差作为目标函数,将每个簇的质点与簇内样本点的平方距离误差和称为畸变程度(distortions),对于一个簇,它的畸变程度越低,代表簇内成员越紧密,畸变程度越高,代表簇内结构越松散。畸变程度会随着类别的增加而降低,但对于有一定区分度的数据,在达到某个临界点时畸变程度会得到极大改善,之后缓慢下降,这个临界点就可以考虑为聚类性能较好的点,从统计的结果来看,选择2聚类比合适。请参见图4,图4为本技术实施例所提供的一种肘部法确定最佳聚类个数的示意图,图4纵坐标score表示畸变程度值,横坐标number clusters表示聚类个数,e表示科学计数法,图4表示在选择不同聚类个数时计算的得到的畸变程度值。
[0137]
步骤d5:阈值确定;
[0138]
根据步骤d4获取的模型,基于每个样本的特征值计算其到聚类中心的欧氏距离。设定一个异常值的比例为1%,因为在标准正太分布的情况下(n(0,1))我们一般认定3个标准差以外的数据为异常值,3个标准差以内的数据包含了数据集中99%以上的数据,所以剩下的1%的数据可以视为异常值;根据所有样本计算出来的欧氏距离和异常值比例计算确定阈值为81.18;
[0139]
步骤d6:离群样本检测,主要通过以下步骤进行;
[0140]
计算临床检测样本的特征值,利用d步骤获取的模型预测临床检测样本的聚类中心,计算该样本到聚类中心的欧氏距离,如果该样本的欧氏距离大于81.18则认为该临床样本异常。
[0141]
步骤d7:异常样本可视化。
[0142]
请参见图5,图5为本技术实施例所提供的一种异常样本可视化示意图,图5的纵坐标distance表示欧氏距离,sample index表示样本索引,如图5所示样本索引ms22050748p1和ms20041621p1为异常样本,anomaly1用于对异常样本进行标记,0表示正常,1表示异常。
[0143]
上述实施例的输入文件包括对照样本和检测样本bam文件,基因组注释文件;使用的软件包括根据位置信息提取映射文件的reads软件,聚类软件;输出的信息包括:不同基因的5
‘
端和3’端位置信息文件,不同基因的5
‘
端和3’测序深度文件,每个样本的欧氏距离文件。
[0144]
当一个样本进行多种测序策略时,需要对样进行一致性分析,当确认样本一致性后才能继续后面的分析。作为一种可行的实施方式,还可以提供以下方式进行数据一致性分析:获取待检测样本的rna fastq数据和dna fastq数据;利用公开软件计算不同数据之间的一致性,命令如下:
[0145]
python ncm_fastq.py-l sample.list-o fastq_check-n matched_list-p 4-pt snp.pt;
[0146]
参数说明如下:
[0147]-l表示样本数据输入文件,示例如下:
[0148]
/data/lsj_r1.fastq
[0149]
/data/lsj_r2.fastq lsj
[0150]
/data/lsh_r1.fastq
[0151]
/data/lsh_r2.fastq lsh
[0152]-o表示选择的数据类型,本实施例用的是fastq数据,所以选择fastq_check;
[0153]-n表示输出文件的前缀;
[0154]-p表示软件所用的线程数;
[0155]-pt表示软件自带的数据库文件;
[0156]
根据最后计算的结果文件判断样本是否一致,如果显示match则表明两个数据据是同一个样本。
[0157]
上述不同数据指:一个样本进行多种测序策略时得到的不同测序数据,比如同一个样本测了dna和rna数据。
[0158]
在得到比对率、所述rrna含量、所述globin rna含量、所述基因3’端测序深度、基因5’端测序深度、以及所述一致性检验结果之后,本实施可以通过以下方式判断该样本是否合格:
[0159]
如果样本比对率小于90%初步判断该样本可能存在其它物种的污染,该样本判断为不合格。
[0160]
如果样本的rrna含量大于0.133或globinrna含量大于0.132,说明rrna或/和globin rna含量异常,可能导致低表达基因因有效数据量低而无法检测的该基因的异常信号,若最终有效数据量小于12g,则需重新建库。
[0161]
基于5’端和3’端测序深度的异常检测,计算临床检测样本特征到聚类中心的欧式距离,如果大于81.1,说明该5’端和3’测序比例显著高于对照群体,可能为rna质量异常或建库异常导致,建议重新取样或者建库。
[0162]
如果一致性检验显示多种测序数据来源一致,可以进行后续的数据分析,如果不一致需要进一步核查原因,确定补救方案。
[0163]
进一步的,本实施例还可以通过以下方式生成所述临床检测样本的质控结果:计算所述比对率对应的第一得分;计算所述rrna含量对应的第二得分;计算所述globin rna含量对应的第三得分;计算所述基因3’端测序深度和基因5’端测序深度对应的第四得分;计算所述一致性检验结果对应的第五得分;将所述第一得分、所述第二得分、所述第三得分、所述第四得分和所述第五得分的总和设置为样本总得分,根据所述样本总得分生成所述临床检测样本的质控结果。
[0164]
上述第一得分根据比对率与标准比对率的偏差程度确定,上述第二得分根据rrna含量与标准rrna含量的偏差程度确定,上述第三得分根据globin rna含量与标准globin rna含量的偏差程度确定,上述第四得分根据基因3’端测序深度与标准基因3’端测序深度的偏差程度、以及基因5’端测序深度与标准基因5’端测序深度的偏差程度确定,上述第五得分根据一致性检验结果与标准一致性检验结果的偏差程度确定,上述偏差程度越大则得分越低,当样本总得分大于预设总得分时判定合格,当样本总得分小于或等于预设总得分时判定不合格。
[0165]
下面通过在实际应用中的实施例说明上述实施例描述的流程。
[0166]
rnaseq已成为依赖高通量测序(hts)技术的转录组分析的常规方法,该技术在转
录水平上提供了比微阵列和其他传统基因表达分析方法更深刻和精确的测量。可用于鉴定新转录本、选择性剪接和基因融合事件、分析等位特异性表达和基因差异表达分析等。然而,由于rna测序技术固有的局限性,质量问题在原始rna序列数据中非常常见。除了各种高通量测序数据中普遍存在的质量问题如测序数据质量(reads数、长度分布、碱基质量、序列复杂度、gc含量等)和来自其他物种的污染外,rnaseq也存一些“rna序列特异性”质量问题,如核糖体rna(rrna)残留、rna降解和不同序列的覆盖度差异。因此,在下游分析之前rna测序数据必须根据实验目的采用严格质量控制(qc),保证结果的可信度。
[0167]
高通量基因组测序在遗传病诊断中的广泛应用使得临床意义未明(vus)变异不断增加,根据acmg指南,vus不能作为临床决策的基础,需要更多的证据来检测这些变异的功能影响。rna测序(rna-seq)作为基因组测序的有效补充方法,可用来解释vus变异的意义,挖掘解析可能的转录水平上的异常,已广泛应用到各种孟德尔疾病的分子诊断(可提高7.5-36%诊断率)。进行rna数据分析时,需评估rna介导的所有事件(可变剪接,基因表达,基因融合,ase等),从而得到全面的遗传信息机制,进一步获得基因诊断。rna-seq技术已逐渐在特定组织和疾病中表现出诊断价值,如用于未确诊神经肌肉病患者的肌肉活检组织样本和线粒体疾病患者来源的成纤维细胞检测等。然而,不管是患者的疾病相关组织还是培养的成纤维细胞都较难获得,外周血作为较易获取的临床样本,使用血液组织rna-seq结合wes/wgs分析,获得不同程度的分子诊断率的提高。
[0168]
外周血是一种非常容易获得的生物流体,提供了关于人体生理和健康状况的丰富信息来源。真核生物细胞含有一些高丰度rna,其中含量高的是核糖体rna(rrna),而血液的rna中含量第二高的就是珠蛋白(globin)rna。这两种rna占血液样本总rna的85%以上。然而,对于用血液样本rna测序(rna-seq)技术进行罕见病遗传病诊断而言,如果不进行处理就建库测序,高丰度的rna占据这大量的数据,不仅浪费了测序成本,也掩藏了目标rna的数据,尤其是一些低丰度但发挥关键作用的患者疾病相关的靶标基因的表达和罕见的剪接异常等,将难以发现显著性的异常。从而影响临床诊断的灵敏性。对于血液样本rna测序数据,可通过实验手段在建库过程中去除rrna和globin rna。并通过特殊质控的设计评估实验去除的效果。
[0169]
rna的质量受多因素的影响,其完整性对globin清除步骤以及下游应用的成功至关重要,即使是中等水平的rna降解也会导致捕获试剂在清除球蛋白mrna时的不充分,血液样本的保存、rna纯化的方法,以及残留rna酶的活性都会影响rna的质量,常用rin值来评估(rin值是rna完整性的参数,其比18s:28s更可靠,不受样本浓度的影响,能提示rna的质量,同时能对测序数据做出预期)。一般测序要求rna rin大于8,有些特殊样本和实验要求rin》7,甚至更小值也会进行下游实验。因此,了解rna实验、数据情况对后续结果解析至关重要,尤其对结果异常的原因分析。常规rnaseq测序常使用polya进行mrna富集,将其随机打断,并以片段化的mrna为模板,随机寡核苷酸为引物进行cdna的合成。纯化后的cdna经过末端修复、加a尾连接测序接头,选择合适的片段大小进行扩增获得测序文库。该方法导致3’端的偏好性(rna完整性越差,对距离3’端越远的序列检测能力越差)。对疾病关联基因3’端,5’端外显子上的测序深度的差异分析可以评估rna完整性,rnaseq建库测序的稳定性,尽可能消减实验造成的诊断的假阴性。再者,rnaseq常作为dna检测的补充,当一个样本进行多种检测时,需要确定样本的一致性,保证dna和rna数据来源于同一个体。
[0170]
目前,许多工具可用于高通量测序数据的qc,如fastqc、fastx-toolkit、qc-chain、ngs qc toolkit。然而,其中大多数主要集中在对一般高通量测序数据的调整上,而不是针对特定高通量测序数据rna序列qc问题。尽管一些工具是专门为rna序列数据设计的,但它们无法对血液组织中的血红蛋白mrna、rrna、疾病关联基因末端覆盖度、dna和rna样本一致性进行质控。因此分析人员迫切需要一种新的技术方案,在高通量测序质控的基础上可以针对血液样本的特性进行进一步的质控,从而提升数据的质量,确定数据的有效性,提高数据分析的灵敏度。
[0171]
本实施例提出了针对血液样本rnaseq质控的全新的技术方案,主要通过以下五个方面对数据进行质控:(1)高通量测序基本质控(qc、比对率、数据量等):通过去除低质量数据和接头序列来获取高质量的目标序列;(2)rrna和globin rna含量质控,判建库测序过程中rrna和globin rna去除是否合格;3基因组比对率分析:判断数据是否污染(4)基因5’端和3
‘
端测序深度质控:判断样本mrna的完整性和建库实验的稳定性(5)样本一致性检验:判断是否存在样本混淆。通过一系列的数据质控,全面了解实验和数据的稳健性,确定分析结果的可靠性。
[0172]
本实施例的技术方案整体流程包括如下过程:原始数据质控;映射文件构建;基于比对率的异常样本检测;rrna、globin rna坐标文件构建;基于rrna、globin rna含量的异常样本检测;基于比对率的异常样本检测;基于基因3’端和5’端测序深度差异分析的异常样本检测;数据一致性分析;根据统计结果判断该样本质控是否合格。
[0173]
请参见图6,图6为本技术实施例所提供的一种针对血液样本rnaseq质控的流程图,图中示出了以下过程:
[0174]
(1)对原始rnaseq fastq数据(即,原始fastq数据文件)进行数据质控,得到质控后的fastq数据(即,目标fastq数据文件);
[0175]
(2)利用基因组序列文件生成索引文件,根据索引文件和质控后的fastq数据进行序列比对生成bam文件;
[0176]
(3)利用bam文件统计比对率;
[0177]
(4)利用基因则序列文件实现rrna和globin rna坐标文件的构建,利用rrna和globin rna坐标文件以及bam文件进行rrna含量统计和globin rna含量统计;
[0178]
(5)判断质控后的fastq数据是否同时包含dna数据和rna数据以便进行数据一致性检验;
[0179]
(6)利用基因组注释文件生成基因3’端和5’端坐标文件,利用所述bam文件进行基因3’端和5’端测序深度统计;
[0180]
(7)基于比对率、rrna含量统计、globin rna含量统计、一致性检验结果和基因3’端和5’端测序深度统计结果综合判断数据质控是否合格。
[0181]
常规的rnaseq测序只会对数据的质量进行质控,缺少了针对不同样本类型特殊性的质控过程,血液中高水平的globin rna和rrna可以占据宝贵的测序空间,影响正常rnaseq分析的检测和定量,从而影响临床诊断的灵敏性。本实施例通过统计globin rna和rrna含量来判断本建库测序过程中globin rna和rrna去除是否合格。
[0182]
rna质量异常或建库异常导致会导致rnaseq测序过程中5’端和3
‘
端测序不均,造成诊断的假阴性。本实施例通过基因5
‘
端和3’端的测序深度的异常检测来判断rna质量或
建库是否存在异常。在rnaseq测序过程中由于各种原因通常会造成数据的污染,本实施例通过统计比对率来判断样本是否污染。在测序中经常会造成样本混淆的问题,本发明通过对数据一致性检验来判断样本是否一致,从而减少了不必要的损失。本实施例所设计的处理流程,对服务器计算资源要求较低,一台普通的8核心64g内存的服务器,能够允许同时运行几十个目标基因的处理任务。本实施例的流程部署简单,使用操作方便,只需部署相关计算节点即可完成全流程分析。
[0183]
请参见图7,图7为本技术实施例所提供的一种临床检测样本的质控装置的结构示意图;
[0184]
该装置可以包括:
[0185]
原始数据质控模块701,用于对临床检测样本进行rna测序获得原始fastq数据文件,对所述原始fastq数据文件进行质控过滤得到目标fastq数据文件;
[0186]
映射文件构建模块702,用于将所述目标fastq数据文件与参考基因组序列文件进行比对,得到bam文件;
[0187]
比对率检测模块703,用于根据所述bam文件确定比对率;
[0188]
目标rna含量统计模块704,用于根据所述bam文件确定rrna含量和globin rna含量;
[0189]
测序深度统计模块705,用于利用所述bam文件进行测序深度检测,得到基因3’端测序深度和基因5’端测序深度;
[0190]
一致性检验模块706,用于对所述临床检测样本进行多种测序策略得到的多种测序数据进行一致性检验,得到一致性检验结果;
[0191]
判断模块707,用于根据所述比对率、所述rrna含量、所述globin rna含量、所述基因3’端测序深度、所述基因5’端测序深度、以及所述一致性检验结果生成所述临床检测样本的质控结果。
[0192]
本实施例对临床检测样本对应的原始fastq数据文件进行质控过滤并与参考基因组序列文件进行比对,得到bam文件,根据上述bam文件可以确定比对率、rrna含量和globin rna含量。本实施例还利用对照血液样本和基因组注释文件对上述bam文件进行测序深度比对,得到临床检测样本的基因3’端测序深度和基因5’端测序深度。本实施例还对所述临床检测样本进行多种测序策略得到的多种测序数据进行一致性检验,得到一致性检验结果。本实施例将上述比对率、rrna含量、globin rna含量、基因3’端测序深度、基因5’端测序深度、以及所述一致性检验结果作为统计指标,综合判断临床检测样本是否合格。上述过程从多个方面对临床检测样本的质量进行检测,结合多个统计指标综合判断临床检测样本是否合格,因此本实施例能够对临床检测样本的质量进行准确检测,提高数据的有效性。
[0193]
进一步的,原始数据质控模块701对所述原始fastq数据文件进行质控过滤得到目标fastq数据文件的过程包括:去除所述原始fastq数据文件中的目标接头序列和低质量序列,得到所述目标fastq数据文件;其中,所述低质量序列包括质量值低于质量值阈值的序列和长度小于长度阈值的序列。
[0194]
进一步的,映射文件构建模块702将所述目标fastq数据文件与参考基因组序列文件进行比对,得到bam文件的过程包括:获取fasta格式的所述参考基因组序列文件,创建所述参考基因组序列文件的索引序列,将所述目标fastq数据文件与所述索引序列进行比对,
得到所述bam文件。
[0195]
进一步的,比对率检测模块703包括:根据所述bam文件确定比对到所述参考基因组序列文件的目标序列;将所述目标序列在所述目标fastq数据文件中所有序列的比例设置为所述比对率。
[0196]
进一步的,还包括:
[0197]
比对率判断模块,用于在根据所述bam文件确定比对率之后,若所述比对率小于第一阈值,则生成判定所述临床检测样本不合格的质控结果。
[0198]
进一步的,还包括:
[0199]
在根据所述bam文件确定rrna含量和globin rna含量之前,目标rna含量统计模块704执行的操作还包括:确定所述参考基因组序列文件中的rrna序列坐标和globin rna序列坐标;按照所述rrna序列坐标和所述globin rna序列坐标统计n个对照血液样本中的对照rrna含量和对照globin rna含量;根据n个所述对照血液样本的对照rrna含量确定第一标准含量区间;根据n个所述对照血液样本的对照globin rna含量确定第二标准含量区间;
[0200]
相应的,在根据所述bam文件确定rrna含量和globin rna含量之后,目标rna含量统计模块704执行的操作还包括:若所述rrna含量不在所述第一标准含量区间内,则生成判定所述临床检测样本不合格的质控结果;若所述globin rna含量不在所述第二标准含量区间内,则生成判定所述临床检测样本不合格的质控结果。
[0201]
进一步的,在利用对照血液样本和基因组注释文件对所述bam文件进行测序深度比对之前,测序深度统计模块705实现的操作还包括:利用基因组注释文件获取基因3’端位置信息和基因5’端位置信息;根据所述基因3’端位置信息确定m个对照血液样本中的基因3’端平均测序深度,并根据所述基因5’端位置信息确定m个所述对照血液样本中的基因5’端平均测序深度;将同一基因对应的基因3’端平均测序深度与基因5’端平均测序深度的比值设置为特征值;利用kmeans算法对所述特征值进行建模得到聚类模型,并根据肘部法确定所述聚类模型的聚类个数;根据所述聚类模型确定距离阈值;
[0202]
相应的,利用所述bam文件进行测序深度检测,得到基因3’端测序深度和基因5’端测序深度之后,测序深度统计模块705实现的操作还包括:将所述基因3’端测序深度和所述基因5’端测序深度的比值设置为样本特征值;计算所述样本特征值与所述聚类模型的聚类中心的欧氏距离;若所述欧式距离大于所述距离阈值,则生成判定所述临床检测样本不合格的质控结果。
[0203]
进一步的,判断模块707根据所述比对率、所述rrna含量、所述globin rna含量、所述基因3’端测序深度、基因5’端测序深度、以及所述一致性检验结果生成所述临床检测样本的质控结果的过程包括:计算所述比对率对应的第一得分;计算所述rrna含量对应的第二得分;计算所述globin rna含量对应的第三得分;计算所述基因3’端测序深度和基因5’端测序深度对应的第四得分;计算所述一致性检验结果对应的第五得分;将所述第一得分、所述第二得分、所述第三得分、所述第四得分和所述第五得分的总和设置为样本总得分,根据所述样本总得分生成所述临床检测样本的质控结果。
[0204]
由于装置部分的实施例与方法部分的实施例相互对应,因此装置部分的实施例请参见方法部分的实施例的描述,这里暂不赘述。
[0205]
本技术还提供了一种存储介质,其上存有计算机程序,该计算机程序被执行时可
以实现上述实施例所提供的步骤。该存储介质可以包括:u盘、移动硬盘、只读存储器(read-only memory,rom)、随机存取存储器(random access memory,ram)、磁碟或者光盘等各种可以存储程序代码的介质。
[0206]
本技术还提供了一种电子设备,可以包括存储器和处理器,所述存储器中存有计算机程序,所述处理器调用所述存储器中的计算机程序时,可以实现上述实施例所提供的步骤。当然所述电子设备还可以包括各种网络接口,电源等组件。
[0207]
说明书中各个实施例采用递进的方式描述,每个实施例重点说明的都是与其他实施例的不同之处,各个实施例之间相同相似部分互相参见即可。对于实施例公开的装置而言,由于其与实施例公开的方法相对应,所以描述的比较简单,相关之处参见方法部分说明即可。应当指出,对于本技术领域的普通技术人员来说,在不脱离本技术原理的前提下,还可以对本技术进行若干改进和修饰,这些改进和修饰也落入本技术权利要求的保护范围内。
[0208]
还需要说明的是,在本说明书中,诸如第一和第二等之类的关系术语仅仅用来将一个实体或者操作与另一个实体或操作区分开来,而不一定要求或者暗示这些实体或操作之间存在任何这种实际的关系或者顺序。而且,术语“包括”、“包含”或者其任何其他变体意在涵盖非排他性的包含,从而使得包括一系列要素的过程、方法、物品或者设备不仅包括那些要素,而且还包括没有明确列出的其他要素,或者是还包括为这种过程、方法、物品或者设备所固有的要素。在没有更多限制的状况下,由语句“包括一个
……”
限定的要素,并不排除在包括所述要素的过程、方法、物品或者设备中还存在另外的相同要素。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1