具有CD3和CD19双重特异性的聚乙二醇化T细胞衔接器的制作方法
具有cd3和cd19双重特异性的聚乙二醇化t细胞衔接器
1.本国际专利申请要求于2021年3月19日提交的国际专利申请号:pct/cn2021/081765的权益,其全部内容通过引用并入以用于所有目的。
技术领域
2.本发明涉及对t细胞上的cd3和b细胞上的cd19具有双重亲和力的t-bsab(t细胞衔接双特异性抗体)及其在治疗自身免疫性疾病中的用途。具体而言,本发明涉及对cd3和cd19具有双重亲和力的聚乙二醇化t-bsab及其在治疗多发性硬化症(ms)以及其它自身免疫性疾病中的用途。
3.发明背景
4.存在超过80种不同的自身免疫性疾病。遗传、饮食、感染和化学品暴露可以涉及在内(campbell,a.w.2014,autoimmune dis 2014,152428)。b细胞通过分泌破坏自身组织的自身抗体、呈递抗原、激活促炎性t细胞和产生细胞因子,在自身免疫性疾病的发病机制中发挥关键作用(sabatino,j.j.等人2019,nat rev neurosci 20,728-745;lee,d.s.w.等人2020,nat rev drug discov,1-21)。b细胞耗竭已被证明是自身免疫性疾病的有效治疗策略(hofmann,k.等人2018,front immunol 9,835)。fda批准的b细胞耗竭疗法已扩展到多种自身免疫性疾病,从器官特异性疾病如天疱疮和ms,到全身性疾病如anca相关性血管炎、类风湿性关节炎(ra)和系统性红斑狼疮(sle)(barnas,j.l.,等人2019,curr opin immunol 61,92-99)。这些疗法主要是基于单克隆抗体的,并靶向具有表面标志物如cd20和cd19的b细胞(townsend,m.j.,等人2010,immunological reviews 237,264-283;frampton,j.e.2020,drugs 80,1259-1264)。然而,抗cd20抗体(奥瑞珠单抗(ocrelizumab)、奥法木单抗(ofatumumab)、利妥昔单抗(rituximab))不能有效去除对传统的免疫抑制疗法极其难治的分泌自身抗体的长寿命浆细胞(chen,d.等人2016,the journal of immunology 196,1541-1549)。事实上,约20%接受利妥昔单抗治疗的ms患者在48周时经历了疾病复发(stephen l.hauser,2008,the new engl and journal of medicine 358,12)。抗cd20抗体疗法未能充分消除的长寿命自身反应性浆细胞可能在一部分ms患者中导致疾病复发。据报道,cd19
+
b细胞在多发性硬化的发病机制中起更关键的作用,抗cd19单克隆抗体在治疗ms的eae(实验性自身免疫性脑脊髓炎)动物模型中显示出比抗cd20抗体更好的治疗效果(chen,d.等人2016,the journal of immunology 196,1541-1549)。然而,抗cd19抗体靶向并耗竭几乎所有b细胞亚群,这可能对患者造成病毒感染的严重威胁。最近的研究表明,接受b细胞耗竭剂治疗的患者经历更高的病毒感染风险、更高的严重疾病率和死亡率(loarce-martos,j.等人2020,rheumatol int 40,2015-2021)。
5.由于并非所有cd19
+
b细胞都参与自身免疫发病机制,因此靶向病理特异性b细胞而不是耗竭所有cd19
+
b细胞是治疗自身免疫性疾病如ms的更有效、更有前景和更安全的疗法。
6.一些研究人员提出,博纳吐单抗(blinatumomab)作为通过将t细胞重新定向到cd19
+
b细胞以裂解b细胞被批准用于all(急性淋巴细胞白血病)治疗的t-bsab或bite(双特
异性t细胞衔接器),可能是自身免疫性疾病治疗的候选药物(musette,p.等人2018,front immunol 9,622)。然而,临床研究显示在接受博纳吐单抗治疗的患者中频繁出现细胞因子释放综合征(crs)和中枢神经系统(cns)毒性。有研究提出,博纳吐单抗的神经毒性可能是由炎性细胞因子的快速释放引起的(topp,m.s.等人2012,blood 120,670-670;portell,c.a.,等人2013,clin pharmacol 5,5-11;bargou,r.等人2008,science 321,974-977;topp,m.s.等人2011,j clin oncol 29 2493-2498;topp,m.s.等人2012,blood 120,5185-5187)。可以预见的是,治疗慢性疾病的药物的安全性要求比抗肿瘤药物更严格。由于自身免疫性疾病是慢性疾病,频繁且严重的crs和cns毒性构成了使用博纳吐单抗治疗自身免疫性疾病的障碍。此外,由于其1.25小时的短循环半衰期,博纳吐单抗通过便携式微型泵经由连续静脉输注施用,这除了与长时间连续输注相关的高感染风险外,还需要癌症患者住院(对自身免疫性疾病患者的治疗是一个挑战)(portell,c.a.,等人2013,clin pharmacol 5,5-11;topp,m.s.等人2012,blood 120,5185-5187)
1,2
。
7.尽管博纳吐单抗在治疗血液系统癌症方面取得了巨大成功,但上述问题和挑战促使开发具有较弱t细胞结合亲和力的t-bsab,以将严重的crs毒性与强大的细胞毒性解耦,并在仍有效激活细胞毒性t细胞以及与病理靶细胞形成免疫突触以杀伤靶细胞的同时最小化细胞因子释放。
8.发明概述
9.在一个方面,本发明提供了治疗自身免疫性疾病的方法,其包括:
10.向受试者施用有效量的式ia化合物或其药学上可接受的盐
[0011][0012]
其中:
[0013]
p是非免疫原性聚合物;
[0014]
t是多官能(例如三官能)小分子接头部分并且具有一个、两个或更多个能够与一个、两个或更多个相同或不同的多肽进行位点特异性缀合的官能团;
[0015]
a1和a2各自独立地选自抗体,例如单链抗体(例如单链可变片段,scfv)、单结构域抗体(纳米抗体)或fab,其中a1和a2之一识别并结合抗原cd3,另一个识别并结合抗原cd19。识别并结合抗原cd19的抗体或fab可以结合cd19的细胞外部分。识别并结合抗原cd3的抗体或fab可以结合t细胞受体复合物中的任何一种cd3复合物亚基,即cd3γ、cd3δ、cd3ε、cd3ζ和cd3η(kuhns,m.s.,等人2006,immunity 24,133-139;2008,r.m.,.plos one 3,e1747)。
[0016]
本发明的另一个方面提供治疗自身免疫性疾病的方法,其包括向受试者施用有效量的式ib化合物或其药学上可接受的盐:
[0017]
[0018]
其中:
[0019]
p是非免疫原性聚合物;
[0020]
b是h或选自c
1-20
烷基和芳基例如c
1-10
烷基和芳基的封端基团,其中所述烷基或芳基的一个或多个碳任选地被杂原子替代;
[0021]
t是具有一个、两个或更多个官能团的三官能(例如氨基酸)接头,其在由双官能间隔物衍生和/或延伸后能够与a1和a2或其衍生物位点特异性缀合,其中t和(l1)a之间的连接基以及t和(l2)b之间的连接基可以相同或不同。
[0022]
l1和l2各自独立地是双官能接头(例如,肽);
[0023]
a和b各自是选自1、2、3、4、5、6、7、8、9和10的整数;
[0024]
a1和a2各自独立地选自抗体,例如单链抗体(例如单链可变片段,scfv)、单结构域抗体(纳米抗体)或fab,其中a1和a2之一识别并结合抗原cd3,另一个识别并结合抗原cd19;
[0025]
和
[0026]
y是选自1-10的整数,例如选自1、2、3、4、5、6、7、8、9和10的整数。
[0027]
在一个方面,识别和结合抗原cd19的抗体或fab可以结合cd19的细胞外部分。在一些实施方案中,识别和结合抗原cd19的抗体是抗cd19 scfv。
[0028]
在一些实施方案中,抗cd19 scfv具有以下氨基酸序列:
[0029][0030]
在本发明的另一个方面,识别和结合抗原cd3的抗体或fab可以结合t细胞受体复合物中的任何一种cd3复合物亚基,即cd3γ、cd3δ、cd3ε和cd3ζη。在一些实施方案中,识别和结合抗原cd3的抗体是抗cd3scfv。
[0031]
在一些实施方案中,抗cd3 scfv具有以下氨基酸序列:
[0032][0033]
非免疫原性聚合物可以选自聚乙二醇(peg)、右旋糖酐、碳水化合物基聚合物、聚环氧烷、聚乙烯醇、羟丙基-甲基丙烯酰胺(hpma)及其共聚物。优选地,非免疫原性聚合物是peg,如支链peg或直链peg。在一些实施方案中,直链peg或支链peg的至少一个末端被h、甲基或低分子量烷基封端。peg的总分子量可以为3,000至100,000道尔顿,例如5,000至80,000、10,000至60,000或20,000至40,000道尔顿。peg可以通过永久键或可裂解键与三官能接头t部分连接。
[0034]
在(l1)a或(l2)b内、(l1)a和蛋白a1之间、(l2)b和蛋白a2之间、t和l1之间或t和l2之间形成连接基的官能团(例如,用于位点特异性缀合的官能团)可以选自卤代烷、酰卤、醛、酮、酯、酸酐、羧酸、酰胺、胺、酰肼、烷基肼、羟基、环氧化物、硫醇、马来酰亚胺、2-吡啶基二硫基变体、芳族砜或乙烯基砜、丙烯酸酯、溴代或碘代乙酰胺、叠氮化物、炔烃、二苯并环辛炔(dbco)、羰基、2-氨基苯甲醛或2-氨基乙酰苯基团、酰肼、肟、酰基三氟硼酸钾、o-氨基甲酰
基羟胺、反式环辛烯、四嗪、三芳基膦等。
[0035]
在一些实施方案中,(l1)a和(l2)b各自可以独立地包含由叠氮化物和炔烃形成或由马来酰亚胺和硫醇形成的连接基。在其他实施方案中,(l1)a、(l2)b和t各自可以独立地是氨基酸或具有2-50个氨基酸单元的肽。在一些实例中,炔烃可以是二苯并环辛炔(dbco)。
[0036]
在一些其他实施方案中,t是赖氨酸,p是peg,并且y是1,而炔烃是二苯并环辛炔(dbco)。
[0037]
在一些实施方案中,a1和a2之一可以衍生自叠氮化物标记的抗体、抗体链、抗体片段、单链抗体或单结构域抗体,其中叠氮化物与(l1)a或(l2)b中各自的炔基缀合;a1和a2中的另一个可以衍生自硫醇标记的抗体、抗体链、抗体片段、单链抗体或单结构域抗体,其中硫醇与(l1)a或(l2)b中各自的马来酰亚胺缀合。
[0038]
上述分子或化合物可以根据包括以下的方法制备:(i)制备具有末端双官能团的非免疫原性聚合物,所述双官能团能够与两种不同的多肽(例如两种不同的抗体)或其修饰形式进行位点特异性缀合;(ii)将非免疫原性聚合物与抗cd3抗体(或其抗原结合片段)和抗cd19抗体(或其抗原结合片段)或它们的修饰形式逐步进行位点特异性缀合以形成式ia或ib的化合物。
[0039]
替代性地,上述聚乙二醇化的t-bsab分子或化合物可以根据包括以下的方法制备:制备具有硫醇标签的抗cd3和抗cd19融合蛋白,然后用硫醇特异性peg试剂如peg马来酰亚胺对融合蛋白进行聚乙二醇化。
[0040]
用本文所述的方法和化合物治疗的自身免疫性疾病包括天疱疮、视神经脊髓炎/视神经脊髓炎谱系障碍(nod/nmod)、多发性硬化症(ms)、anca相关性血管炎、类风湿性关节炎(ra)、克罗恩病、炎症性肠病(ibd)和系统性红斑狼疮(sle)、哮喘、银屑病/银屑病关节炎、艾迪生病、格雷夫斯病、特应性皮炎、红斑狼疮、1型糖尿病等。上述列表并非排他性的,普通技术人员将认识到本文未具体提及的其他自身免疫性疾病旨在包括在内。
[0041]
在一些实施方案中,待治疗的自身免疫性疾病是ms。在一些实施方案中,该疾病是对使用如以下的化合物的常规免疫抑制疗法表现出耐药性或难治性的ms:tecfidera、芬戈莫德(gilenya)、tysabri、奥巴捷(aubagio)、克拉屈滨(mavenclad)、克帕松(copaxone)、ifn-β-1a、ifn-β-1b、抗cd52抗体(阿仑珠单抗(alemtuzumab)、阿仑珠单抗(alemtuzumab))、那他珠单抗(natalizumab)、抗cd19药剂(伊奈利珠单抗(inebilizumab)、obexelimab)或抗cd20药剂(利妥昔单抗、奥瑞珠单抗、奥法木单抗)。
[0042]
在一些实施方案中,待治疗的疾病是对与b细胞耗竭剂如抗cd19或抗cd20单克隆抗体的施用相关的疗法表现出耐药性或难治性的自身免疫性疾病。在其他实施方案中,待治疗的疾病是对与抗cd3x抗cd20双特异性抗体的施用相关的疗法表现出耐药性或难治性的自身免疫性疾病。
[0043]
在一些实施方案中,式ia或ib的化合物或其药学上可接受的盐在自身免疫性疾病的治疗开始时(例如作为一线疗法)或在随后的治疗轮次中(例如作为二线、三线或四线疗法)施用于受试者。在一些实施方案中,自身免疫性疾病是ms。在一些实施方案中,该化合物对停止治疗后复发的ms的治疗有效。
[0044]
在一些实施方案中,式ia或ib的化合物或其药学上可接受的盐以0.05mg/kg/剂至50mg/kg/剂的量施用。在一些实施方案中,对于每个治疗周期,每4-8周施用本文所述的化
合物一次至八次,或在4-8周内施用一次至四次,随后每个周期给予一周的休息期,直到证明有期望的结果。
[0045]
在一些实施方案中,有效量的化合物与用于治疗自身免疫性疾病的另一种治疗剂同时或依次施用。
[0046]
本方法和化合物的一个优点是本文所述的聚乙二醇化抗cd3x抗cd19t-bsab具有降低的毒性和/或克服现有药剂遇到的问题。
[0047]
在另一个方面,本发明提供了组合物,例如药物组合物,其中该组合物包含式ia或ib的化合物或其药学上可接受的盐,以及任选地药学上可接受的载剂、赋形剂或稀释剂。
[0048]
在一些实施方案中,组合物还包含用于治疗自身免疫性疾病的第二治疗剂。在一些实施方案中,第二治疗剂可以选自小分子药剂如tecfidera、芬戈莫德、tysabri、奥巴捷、mavenclad;肽制剂如克帕松(copaxone)、蛋白质生物制剂如ifn-β-1a、ifn-β-1b、抗cd52抗体(阿仑珠单抗,alemtuzumab)、那他珠单抗;b细胞耗竭剂如抗cd19药剂(伊奈利珠单抗、obexelimab)、抗cd20药剂(利妥昔单抗、奥瑞珠单抗、奥法木单抗)。
[0049]
在一个方面,本发明提供式ia、ib的化合物或其药学上可接受的盐或上述组合物在制备用于预防、治疗或减轻受试者的自身免疫性疾病(例如ms)的药物中的用途。
[0050]
在另一个方面,本发明提供上述式ia或ib的化合物或其药学上可接受的盐或组合物,其用于预防、治疗或减轻受试者的自身免疫性疾病(例如ms)。
[0051]
本发明的一个或多个实施方案的细节在以下描述和附图中阐述。本发明的其他特征、目的和优点将从说明书和权利要求中显而易见。
附图说明
[0052]
图1:实施例1中elisa测定比较与人cd19或人cd3e&cd3d蛋白的结合亲和力。
[0053]
图2:实施例2中未观察到jy108对cd19-细胞的药物特异性细胞毒性。
[0054]
图3:实施例2中jy108对pbmc中b细胞的消除。
[0055]
图4:实施例2中e:t比率对jy108功效的影响。
[0056]
图5:实施例2中细胞系对jy108功效的影响。
[0057]
图6:实施例3中jy108对前-b all肿瘤模型的体内功效。
[0058]
图7:实施例4中jy108的药代动力学特性。
[0059]
图8:实施例5中jy108与转基因动物模型中靶标的结合。
[0060]
图9:实施例5中来自转基因小鼠(hcd3hcd19)的脾细胞作为jy108的效应物。
[0061]
图10:实施例6中jy108在10mg/kg的单剂量下没有急性毒性。
[0062]
图11:实施例7中由jy108导致的从人全血中释放的细胞因子较少。
[0063]
图12:实施例8中jy108显著改善了eae症状。
[0064]
图13:实施例8中组织病理学切片的lfb染色。
[0065]
图14:实施例8中由jy108导致的mog特异性自身抗体的耗竭。
[0066]
图15:实施例9中jy108与medi-551的头对头比较。
[0067]
图16:实施例10中由jy108导致的cd19
+
b细胞的部分去除。
[0068]
图17:实施例10中b
reg
细胞对medi-551和jy108都有耐药性。
[0069]
发明详述
[0070]
在本发明中,提供了使用对t细胞的人cd3和b细胞的人cd19具有双重亲和力的聚乙二醇化t-bsab治疗自身免疫性疾病,特别是ms的方法。聚乙二醇化t-bsab能够选择性耗竭表达致病性cd19的b细胞,同时释放最少的细胞因子。
[0071]
a.治疗方法
[0072]
在一个方面,本发明提供了治疗自身免疫性疾病的方法,其包括:
[0073]
向受试者施用有效量的式ia化合物或其药学上可接受的盐,
[0074][0075]
其中:
[0076]
p是非免疫原性聚合物;
[0077]
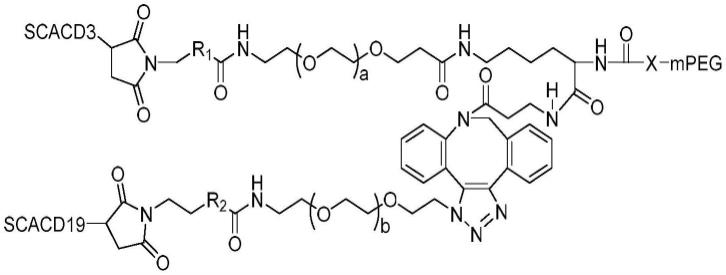
t是多官能(例如三官能)小分子接头部分并且具有一个、两个或更多个能够与一个、两个或更多个相同或不同的多肽位点特异性缀合的官能团;
[0078]
a1和a2各自独立地选自抗体,例如单链抗体(例如单链可变片段,scfv)、单结构域抗体(纳米抗体)或fab,其中a1和a2之一识别并结合抗原cd3,另一个识别并结合抗原cd19。识别并结合抗原cd19的抗体或fab可以结合cd19的细胞外部分。识别并结合cd3的抗体或fab可以结合t细胞受体复合物中的任何一种cd3复合物亚基,即cd3γ、cd3δ、cd3ε和cd3ζη。
[0079]
本发明的另一个方面提供治疗自身免疫性疾病的方法,其包括向受试者施用有效量的式ib化合物或其药学上可接受的盐:
[0080][0081]
其中:
[0082]
p是非免疫原性聚合物;
[0083]
b是h或选自c
1-20
烷基和芳基例如c
1-10
烷基和芳基的封端基团,其中所述烷基或芳基的一个或多个碳任选地被杂原子替代;
[0084]
t是具有一个、两个或更多个官能团的三官能(例如氨基酸)接头,其在由双官能间隔物衍生和/或延伸后能够与a1和a2或其衍生物位点特异性缀合,其中t和(l1)a之间的连接基以及t和(l2)b之间的连接基可以相同或不同。
[0085]
l1和l2各自独立地是双官能接头(例如肽);
[0086]
a和b各自是选自1、2、3、4、5、6、7、8、9和10的整数;
[0087]
a1和a2各自独立地选自抗体,例如单链抗体(例如单链可变片段,scfv)、单结构域抗体(纳米抗体)或fab,其中a1和a2之一识别并结合抗原cd3,另一个识别并结合抗原cd19;和
[0088]
y是选自1、2、3、4、5、6、7、8、9和10的整数。
[0089]
在一个方面,识别和结合抗原cd19的抗体或fab可以结合cd19的细胞外部分。在一些实施方案中,识别和结合抗原cd19的抗体是抗cd19 scfv。
[0090]
在一些实施方案中,抗cd19 scfv具有以下氨基酸序列:
[0091][0092]
在本发明的另一个方面,识别和结合抗原cd3的抗体或fab可以结合t细胞受体复合物中的任何一种cd3复合物亚基,即cd3γ、cd3δ、cd3ε和cd3ζη。在一些实施方案中,识别和结合抗原cd3的抗体是抗cd3scfv。
[0093]
在一些实施方案中,抗cd3 scfv具有以下氨基酸序列:
[0094][0095]
b.聚合物部分p
[0096]
化合物中的非免疫原性聚合物p可以选自聚乙二醇(peg)、右旋糖酐、碳水化合物基聚合物、聚环氧烷、聚乙烯醇、羟丙基-甲基丙烯酰胺(hpma)及其共聚物。优选地,非免疫原性聚合物是peg,如支链peg或直链peg。在一些实施方案中,直链peg或支链peg的至少一个末端被h、甲基或低分子量烷基封端。peg的总分子量可以为3,000至100,000道尔顿,例如5,000至80,000、10,000至60,000或20,000至40,000道尔顿。peg可以通过永久键或可裂解键与三官能接头t部分连接。
[0097]
聚合物可以包含能够被官能化、活化或缀合至反应配偶体的端基。端基的非限制性实例包括羟基、氨基、羧基、巯基和卤素(halide)。
[0098]
在一些实施方案中,y是1并且式ib代表具有悬挂聚合物链的化合物。末端b可以用作封端基团。
[0099]
在一些实施方案中,y是2、3、4、5或6并且式ib代表包含分支的聚合物部分的化合物。在一些实施方案中,[b-p]y中的b是低分子量c
1-10
烷基,如甲基、乙基和丁基,其中一个或多个碳可以被杂原子(例如o、s和n)替代。
[0100]
在本发明的另一个实施方案中,可以使用替代的支链peg。分支的p部分可以衍生自下式化合物:
[0101]
(b-peg)ml-s
i-fi[0102]
其中:
[0103]
peg是聚乙二醇。m是大于1的整数,以优选地提供总分子量为3000至50000道尔顿或更大(如果期望)的聚合物。b是甲基或其他低分子量烷基。l是附接两个或更多个peg的功能连接部分。此类连接部分的实例包括:任何氨基酸,如甘氨酸、丙氨酸、赖氨酸或1,3-二氨基-2-丙醇、三乙醇胺、任何5或6元芳环或具有两个以上附接的官能团的脂环。s是任何不可裂解的间隔物。f是末端官能团如羟基、羧基、巯基、氨基等。i是0或1。
[0104]
当i等于0时,式为:
[0105]
(b-peg)ml
[0106]
其中peg、m、b和l具有与上述相同的定义。
[0107]
c.三官能接头t
[0108]
t代表三官能接头,与p、(l1)a和(l2)b连接。t可以衍生自具有三个官能团的任意组合的分子,官能团的非限制性实例包括羟基、氨基、肼基、羧基、巯基和卤素。三官能接头中的官能团可以相同或不同。三官能接头的一个或多个官能团可以在t和反应配偶体之间的反应之前或之后转化为一个或多个其它基团。例如,羟基可以转化为甲磺酸酯或甲苯磺酸酯基团。卤素可以被叠氮基取代。t的酸性官能团可以通过与带有末端炔烃的氨基偶联转化为炔烃官能团。
[0109]
在一些实施方案中,t可以衍生自选自以下的天然或非天然氨基酸:半胱氨酸、赖氨酸、天冬酰胺、天冬氨酸、谷氨酸、谷氨酰胺、组氨酸、丝氨酸、苏氨酸、色氨酸、酪氨酸或遗传编码的烯烃赖氨酸(如n6-(己-5-烯酰基)-l-赖氨酸)、2-氨基-8-氧代壬酸、间或对乙酰基苯丙氨酸、带有β-二酮侧链的氨基酸(如2-氨基-3-(4-(3-氧代丁酰基)苯基)丙酸)、(s)-2-氨基-6-(((1r,2r)-2-叠氮基环戊基氧基)羰基氨基)己酸、叠氮基高丙氨酸、吡咯赖氨酸类似物n
6-((丙-2-炔-1-基氧基)羰基)-l-赖氨酸、(s)-2-氨基-6-戊-4-炔酰氨基己酸、(s)-2-氨基-6-((丙-2-炔基氧基)羰基氨基)己酸、(s)-2-氨基-6-((2-叠氮基乙氧基)羰基氨基)己酸、对叠氮基苯丙氨酸、对叠氮基苯丙氨酸、n
ε-丙烯酰基-l-赖氨酸、nε-5-降冰片烯-2-基氧基羰基-l-赖氨酸、n-ε-(环辛-2-炔-1-基氧基环辛-2-炔-1-基氧基)羰基)-l-赖氨酸、n-ε-(2-(环辛-2-炔-1-基氧基)乙基)羰基-l-赖氨酸、以及遗传编码的四嗪氨基酸(如4-(6-甲基-s-四嗪-3-基)氨基苯丙氨酸)。
[0110]
d.双官能接头l1和l2[0111]
每个接头部分l1和l2包含接头链、内部连接基和/或末端连接基。接头链可以独立地选自氨基酸或具有2至50个氨基酸残基的肽,或-(ch2)mxy(ch2)
n-、-x(ch2)mo(ch2ch2o)
p
(ch2)ny-、-(ch2)mx-y(ch2)
n-、-(ch2)m杂环基-、-(ch2)mx-、-x(ch2)my-,其中m、n和p在每种情况下独立地为0至25范围的整数;x和y在每种情况下独立地选自c(=o)、cr1r2、nr3、s、o或无,其中r1和r2独立地代表氢、c
1-10
烷基或(ch2)
1-10
c(=o),r3是h或c
1-10
烷基,并且其中杂环基衍生自马来酰亚胺基、应变烯烃和炔烃、叠氮化物或四唑基部分。
[0112]
在一个方面,接头链可以独立地选自:-(ch2)ac(o)nr1(ch2)
b-、-(ch2)ao(ch2ch2o)
c-、-(ch2)a杂环基-、-(ch2)ac(o)-、-(ch2)anr
1-、-cr1=n-nr
1-、-cr1=n-o-、-cr1=n-nr
2-co-、-n=n-co-、-s-s-,其中a、b和c各自是独立地选自0至25的整数,包括所有亚基;r1和r2独立地代表氢或c1-c10烷基。
[0113]
l1和l2(在内部位置或在末端位置)的杂环基连接基团可以衍生自基于马来酰亚胺的部分。合适的前体的非限制性实例包括n-琥珀酰亚胺基4-(马来酰亚胺甲基)环己烷羧酸酯(smcc)、n-琥珀酰亚胺基-4-(n-马来酰亚胺甲基)-环己烷-1-羧基-(6-酰胺基己酸酯)(lc-smcc)、κ-马来酰亚胺基十一烷酸n-琥珀酰亚胺基酯(kmua)、γ-马来酰亚胺基丁酸n-琥珀酰亚胺基酯(gmbs)、ε-马来酰亚胺基己酸n-羟基琥珀酰亚胺基酯(emcs)、间马来酰亚胺基苯甲酰基-n-羟基琥珀酰亚胺酯(mbs)、n-(α-马来酰亚胺基乙酰氧基)-琥珀酰亚胺酯(amas)、琥珀酰亚胺-基6-(β-马来酰亚胺基丙酰胺基)己酸酯(smph)、n-琥珀酰亚胺基4-(对马来酰亚胺基苯基)-丁酸酯(smpb)和n-(对马来酰亚胺基苯基)异氰酸酯(pmpi)。
[0114]
替代性地,l1和l2的杂环基连接基团可以是四唑基、反式环辛烯、叠氮化物或应变炔烃。例如,杂环基三唑基连接基可以由两个不同接头部分的缀合形成:叠氮化物和应变炔
烃。因此,杂环基也可以用作连接点。
[0115]
在一些实施方案中,(l1)a和/或(l2)b包含:
[0116]
x
1-(ch2)ac(o)nr1(ch2)bo(ch2ch2o)c(ch2)dc(o)-或
[0117]
x
3-(ch2)ac(o)nr1(ch2)bo(ch2ch2o)c(ch2)
d x2(ch2)en r2,
[0118]
其中x1、x2和x3可以相同或不同并且独立地代表杂环基;
[0119]
a、b、c、d和e各自是独立地选自1-25的整数;和
[0120]
r1和r2独立地代表氢或c1-c10烷基。
[0121]
在一些实施方案中,x1和/或x3衍生自基于马来酰亚胺的部分。在一些实施方案中,x2代表三唑基。在一些实施方案中,r1和r2各自代表氢。在一些实施方案中,a、b、c、d和e各自是独立地选自0、1、2、3、4、5、6、7、8、9和10的整数。
[0122]
在一些其他非限制性示例性实施方案中,接头部分l1和l2各自还可以衍生自基于卤代乙酰基的部分,该部分选自n-琥珀酰亚胺基-4-(碘乙酰基)-氨基苯甲酸酯(siab)、n-琥珀酰亚胺基碘乙酸酯(sia)、n-琥珀酰亚胺基溴乙酸酯(sba)和n-琥珀酰亚胺基3-(溴乙酰氨基)丙酸酯(sbap)。
[0123]
e.连接基团
[0124]
本发明的化合物或缀合物的不同部分可以通过各种化学连接基连接。实例包括但不限于酰胺、酯、二硫化物、醚、氨基、氨基甲酸酯、肼、硫醚和碳酸酯。
[0125]
例如,可以活化peg部分(p)的末端羟基,然后与赖氨酸(t)偶联以提供式ia或ib的p和t之间的期望连接点。同时,t和l1或l2之间的连接基团可以是由接头l1或l2的氨基与赖氨酸(t)的羧基反应产生的酰胺。替代性地,t和l1或l2之间的连接基团可以是由t的氨基与接头l1或l2的活化羧基反应产生的酰胺。根据化合物或缀合物的期望特性,也可以在抗体部分(a)和相邻接头(l1或l2)之间以及在l1或l2的各个接头之间或之内引入合适的连接基团。
[0126]
在一些实施方案中,化合物或缀合物的不同部分之间的连接基团可以衍生自一对彼此具有固有化学亲和力和选择性的官能团的偶联。这些类型的偶联或成环允许位点特异性缀合以引入特定多肽或抗体部分。导致位点特异性缀合的这些官能团的非限制性实例包括硫醇、马来酰亚胺、2
’‑
吡啶基二硫基变体、芳族或乙烯基砜、丙烯酸酯、溴代或碘代乙酰胺、叠氮化物、炔烃、二苯并环辛炔(dbco)、羰基、2-氨基-苯甲醛或2-氨基-乙酰苯基团、酰肼、肟、酰基三氟硼酸钾、o-氨基甲酰基羟胺、反式-环辛烯、四嗪和三芳基膦。
[0127]
在一些实施方案中,(l1)a或(l2)b各自可以独立地包含由叠氮化物和炔烃形成或由马来酰亚胺和硫醇形成的连接基。在其他实施方案中,(l1)a、(l2)b和t各自可以是氨基酸或具有2-50个氨基酸单元的肽。
[0128]
在一些实施方案中,炔烃可以是二苯并环辛炔(dbco)。在其他情况下,t是赖氨酸,p是peg,并且y是1,而炔烃是二苯并环辛炔(dbco)。在另一个实施方案中,a1和a2之一可以衍生自叠氮化物标记的抗体、抗体链、抗体片段、单链抗体或单结构域抗体,其中叠氮化物与(l1)a或(l2)b中各自的炔烃缀合;a1和a2中的另一个可以衍生自硫醇标记的抗体、抗体链、抗体片段、单链抗体或单结构域抗体,其中硫醇与(l1)a或(l2)b中各自的马来酰亚胺缀合。
[0129]
f.对cd3和cd19具有双重特异性的聚乙二醇化t-bsab的合成
[0130]
在一些实施方案中,p代表peg部分。在一些示例性实施方案中,peg的末端官能团如羟基或羧基等被活化并与三官能小分子部分如boc保护的赖氨酸缀合以形成末端分支的
异双官能peg。然后通过与具有炔基的小分子间隔物偶联,将新形成的羧基转化为炔基。boc去保护后的裸氨基与另一个具有马来酰亚胺基团的小分子间隔物缀合形成末端分支的马来酰亚胺/炔烃异双官能peg。所得马来酰亚胺/炔烃末端分支的异双官能peg连续与硫醇标记的单链抗cd3抗体和叠氮化物标记的单链抗cd19抗体进行位点特异性缀合,以形成聚乙二醇化t-bsab。
[0131]
合成的细节描述于名称为“long acting multi-specific molecules and related methods”的专利申请wo 2018/075308al中,其内容通过整体引用并入本文。
[0132]
g.组合物/制剂
[0133]
本发明还提供组合物,例如药物组合物,其含有本发明的聚乙二醇化的t-bsab分子,任选地与药学上可接受的载剂一起配制。例如,本发明的药物组合物可以包含与cd3和cd19两者结合的聚乙二醇化t-bsab分子。
[0134]
本发明的治疗制剂可以通过将具有期望纯度的聚乙二醇化t-bsab与任选的生理学上可接受的载剂、赋形剂或稳定剂混合来制备(remington'spharmaceutical sciences 1980,第16版,osol,a.ed.),形式为冻干制剂或水溶液。可接受的载剂、赋形剂或稳定剂在所采用的剂量和浓度下对接受者无毒,包括缓冲剂,如磷酸盐、柠檬酸盐和其他有机酸;抗氧化剂,包括抗坏血酸和甲硫氨酸;防腐剂(如十八烷基二甲基苄基氯化铵;六甲铵氯化物;苯扎氯铵、苄索氯铵;苯酚、丁醇或苯甲醇;对羟基苯甲酸烷基酯如对羟基苯甲酸甲酯或对羟基苯甲酸丙酯;邻苯二酚;间苯二酚;环己醇;3-戊醇;和间甲酚);低分子量(少于约10个残基)蛋白质,如血清白蛋白、明胶或免疫球蛋白;亲水性聚合物,如聚乙烯吡咯烷酮;氨基酸,如甘氨酸、谷氨酰胺、天冬酰胺、组氨酸、精氨酸或赖氨酸;单糖、二糖和其他碳水化合物,包括葡萄糖、甘露糖或糊精;螯合剂,如edta;糖,如蔗糖、甘露糖醇、海藻糖或山梨糖醇;成盐抗衡离子,如钠;金属络合物(例如,锌-蛋白质络合物);和/或非离子表面活性剂,如tween、pluronics或聚乙二醇(peg)。
[0135]
该制剂还可以根据待治疗的特定适应症的需要含有一种以上的活性化合物,优选具有互补活性且不会相互产生不利影响的活性化合物。例如,该制剂可以进一步包含另一种抗体或抗自身免疫性疾病剂。此类分子可以适当地以对预期目的有效的数量组合存在。
[0136]
活性成分也可以被包裹在通过例如凝聚技术或界面聚合制备的微胶囊中,例如分别在胶体药物递送系统(例如脂质体、白蛋白微球、微乳液、纳米颗粒和纳米胶囊)或粗乳液中的羟甲基纤维素或明胶微胶囊和聚(甲基丙烯酸甲酯)微胶囊。此类技术在remington's pharmaceutical sciences 1980,第16版,osol,a.ed.中公开。可以制备缓释制剂。缓释制剂的合适实例包括含有聚乙二醇化的t-bsab分子的固体疏水聚合物的半透性基质,该基质是成形制品的形式例如薄膜或微胶囊。可缓释基质的实例包括聚酯、水凝胶(例如,聚(甲基丙烯酸2-羟乙基)或聚(乙烯醇))、聚乳酸(美国专利号3773919)、l-谷氨酸和γ-乙基-l-谷氨酸的共聚物、不可降解的乙烯-乙酸乙烯酯、可降解的乳酸-乙醇酸共聚物如lupron depot(由乳酸-乙醇酸共聚物和乙酸亮丙瑞林组成的可注射微球)和聚-d-(-)-3-羟基丁酸。虽然聚合物如乙烯-乙酸乙烯酯和乳酸-乙醇酸等聚合物能够释放分子超过100天,但某些水凝胶在更短的时间内释放蛋白质。当包封的抗体在体内长时间滞留时,它们可能会因暴露在37℃下的湿度中而发生变性或聚集,从而导致生物活性丧失和免疫原性的可能变化。根据所涉及的机制,可以设计合理的策略以实现稳定化。例如,如果发现聚集机制是通
过硫代二硫化物交换形成分子间s-s键,则可以通过修饰巯基残基、从酸性溶液中冻干、控制水分含量、使用适当的添加剂和开发特定的聚合物基质组合物来实现稳定化。
[0137]
本发明的药物组合物可以在联合疗法中施用,即与其他药剂联合。可用于联合疗法的治疗剂的实例包括:小分子药剂如tecfidera、芬戈莫德、tysabri、奥巴捷、克拉屈滨;肽制剂如克帕松、重组体多肽或蛋白质如ifn-β-1a、ifn-β-1b、抗cd52抗体(阿仑珠单抗,alemtuzumab)、那他珠单抗;b细胞耗竭剂如抗cd19药剂(伊奈利珠单抗、在ii期中的obexelimab)、抗cd20药剂(利妥昔单抗、奥瑞珠单抗、奥法木单抗)。
[0138]
用于体内施用的制剂应该是无菌的。这可以通过无菌过滤膜过滤很容易地实现。无菌可注射溶液可以通过将所需量的活性化合物加入适当的溶剂和上述列举的一种成分或成分的组合(根据需要)中来制备,然后进行灭菌微滤。通常,分散体通过将活性化合物引入无菌载体中来制备,该载体含有基本的分散介质和来自上述列举的那些的所需的其他成分。在用于制备无菌注射溶液的无菌粉末的情况下,优选的制备方法是真空干燥和冷冻干燥(冻干),从其先前无菌过滤的溶液产生活性成分加上任何其他期望成分的粉末。
[0139]
h.剂量
[0140]
可与载剂材料组合以产生单一剂型的活性成分的量将根据待治疗的受试者和特定的施用方式而变化。可与载剂材料组合以产生单一剂型的活性成分的量通常是产生治疗效果的组合物的量。通常,该量的范围为约0.01%至约99%的活性成分,优选地约0.1%至约70%,最优选地为约1%至约30%的活性成分与药学上可接受的载剂组合。
[0141]
调整剂量方案以提供最佳的期望反应(例如,治疗反应)。例如,可以单次施用,在一段时间内分几剂施用,或者根据治疗情况的紧急程度按比例减少或增加剂量。以剂量单位形式配制肠胃外组合物以便于施用和剂量均匀是特别有利的。如本文所用的剂量单位形式是指适合用于待治疗受试者的作为单位剂量的物理上离散的单位:每个单位含有预定量的活性化合物,经计算可与所需的药物载剂一起产生期望的治疗效果。本发明的剂量单位形式的规格取决于并直接依赖于(a)活性化合物的独特特性和待实现的特定治疗效果,和(b)在复合此活性化合物用于个体敏感性治疗的领域中固有的限制。
[0142]
对于本发明的聚乙二醇化t-bsab的施用,剂量范围为约0.0001至100mg/kg,更通常为0.05至50mg/kg宿主体重。例如剂量可以是0.01mg/kg体重、0.1mg/kg体重、1mg/kg体重、5mg/kg体重、10mg/kg体重或50mg/kg体重或在0.1-10mg/kg的范围内。示例性治疗方案需要如下施用:每周两次或三次、每周一次、每两周一次、每三周一次、每四周一次、每月一次、每3个月一次或每三至6个月一次。本发明的聚乙二醇化t-bsab分子的优选给药方案包括通过静脉内施用0.25mg/kg体重或1mg/kg体重,使用以下给药方案之一给予聚乙二醇化t-bsab分子:(i)每四周施用八剂,然后每三个月;(ii)每两周一次;(iii)按1mg/kg体重施用一次,然后每两周按0.25mg/kg体重施用一次。
[0143]
替代性地,聚乙二醇化t-bsab分子可以作为缓释制剂施用,在这种情况下需要较少的施用频率。剂量和频率取决于患者体内聚乙二醇化t-bsab分子的半衰期。通常,用较高分子量peg修饰的t-bsab具有较长的半衰期。施用的剂量和频率可以根据治疗是预防性的还是治疗性的而有所不同。在预防性应用中,在长时间内以相对不频繁的间隔施用相对低的剂量。一些患者在余生中持续接受治疗。在治性疗应用中,有时需要在相对短的间隔时间内使用相对高的剂量,直到疾病的进展减少或终止,优选地直到病人显示出部分或完全的
疾病症状改善。此后,可以向患者施用预防性方案。可以改变本发明药物组合物中活性成分的实际剂量水平,以获得有效实现特定患者、组合物和施用方式的期望治疗反应的活性成分的量。所选剂量水平取决于多种药代动力学因素,包括所用的本发明特定组合物的活性、施用途径、施用时间、所用特定化合物的排泄率、治疗持续时间、与所用特定组合物联用的其他药物、化合物和/或材料、待治疗患者的年龄、性别、重量、病症、总体健康和既往病史、和医学领域熟知的其他因素。
[0144]
本发明的聚乙二醇化t-bsab分子的“治疗有效剂量”优选导致疾病症状的严重程度降低、无疾病症状期的频率和持续时间增加、或由于疾病折磨的损伤或残疾的预防。例如,对于ms的治疗,“治疗有效剂量”优选将临床评分相对于未治疗的受试者降低至少约20%,更优选至少约40%,甚至更优选至少约60%,还更优选至少约80%。可以在预测人类疾病功效的动物模型系统中评估药剂或化合物降低临床评分的能力。替代性地,可以通过本领域技术人员已知的测定法检查化合物在体外耗竭cd19
+
b细胞的能力来评估组合物的这种性质。治疗有效量的治疗化合物可以降低临床评分,或以其他方式改善受试者的症状。本领域普通技术人员将能够基于如受试者的体型、受试者症状的严重性和所选择的特定组合物或施用途径等因素来确定这样的量。
[0145]
i.施用
[0146]
本发明的组合物可以使用本领域已知的多种方法中的一种或多种通过一种或多种施用途径来施用。如本领域技术人员所理解的,施用途径和/或方式将根据期望的结果而变化。本发明的聚乙二醇化t-bsab的优选施用途径包括静脉内、肌肉内、皮内、腹膜内、皮下、脊髓或其他肠胃外施用途径,例如通过注射或输注。如本文所用,短语“肠胃外施用”是指除肠内和局部施用以外的施用方式,通常通过注射,包括但不限于静脉内、肌内、动脉内、鞘内、囊内、眶内、心内、皮内、腹腔内、经气管、皮下、表皮下、关节内、被膜下、蛛网膜下、脊柱内、硬膜外和胸骨内注射和输注。
[0147]
替代性地,本发明的聚乙二醇化t-bsab可以通过非肠道途径施用,如局部、表皮或粘膜施用途径,例如经鼻、经口、经阴道、经直肠、经舌下或经局部。
[0148]
活性化合物可以与载剂一起制备,所述载剂将保护化合物免于快速释放,如控释制剂,包括植入物、透皮贴剂和微囊化递送系统。可以使用可生物降解的、生物相容的聚合物,如乙烯乙酸乙烯酯、聚酐、聚乙醇酸、胶原、聚原酸酯和聚乳酸。许多制备此类制剂的方法已获得专利,或为本领域技术人员普遍所知。参见例如,sustained and controlled release drug delivery systems,1978,j.r.robinson,ed.,marcel dekker,inc.,new york。
[0149]
治疗性组合物可以用本领域已知的医疗装置施用。例如,本发明的治疗性组合物可以用无针皮下注射装置施用,如美国专利号5399163、5383851、5312335、5064413、4941880、4790824和4596556中公开的装置。可用于本发明的众所周知的植入物和组件的实例包括在美国专利号4487603、4486194、4447233、4447224、4439196和4475196中描述的那些。这些专利通过引用并入本文。许多其他这样的植入物、递送系统和组件是本领域技术人员已知的。
[0150]
j.治疗
[0151]
用聚乙二醇化t-bsab治疗的疾病可以是任何自身免疫性疾病,包括但不限于天疱
疮、视神经脊髓炎/视神经脊髓炎疾病(nod/nmod)、多发性硬化症(ms)、anca相关性血管炎、类风湿性关节炎(ra)、克罗恩病、炎症性肠病(ibd)和系统性红斑狼疮(sle)、哮喘、牛皮癣、特应性皮炎、红斑狼疮、1型糖尿病。
[0152]
在本发明的一个方面,自身免疫性疾病是多发性硬化症(ms)。
[0153]
在另一个方面,自身免疫性疾病对如下所列的任何先前疗法是难治性的或耐药性的。对于本发明的目的,难治性或耐药性自身免疫性疾病定义为那些对先前的疗法或治疗没有反应的疾病。自身免疫性疾病可以在治疗开始时是耐药或难治的,或者它们可以在治疗期间变得耐药或难治。难治性自身免疫性疾病包括那些在治疗开始时没有反应或最初在短时间内反应但后来没有反应的疾病。难治性自身免疫性疾病还包括那些对使用一种常规疗法的治疗有反应但对随后的几轮疗法没有反应的疾病。对于本发明的目的,难治性自身免疫性疾病还涵盖那些似乎通过治疗有效但在停止治疗后最多一年内、有时最多五年内或更长时间内复发的疾病。常规疗法可以采用小分子药剂如tecfidera、芬戈莫德、tysabri、奥巴捷、克拉屈滨;肽制剂如克帕松、重组体多肽或蛋白质如ifn-β-1a、ifn-β-1b、抗cd52抗体(阿仑珠单抗,alemtuzumab)、那他珠单抗;b细胞耗竭剂如抗cd19药剂(伊奈利珠单抗、ii期中的obexelimab)、抗cd20药剂(利妥昔单抗、奥瑞珠单抗、奥法木单抗)或其组合。为了便于描述而非限制,应理解难治性自身免疫性疾病可与耐药性自身免疫性疾病互换。
[0154]
对于本发明的目的,耐药性或难治性自身免疫性疾病的成功治疗应理解为与在没有本文所述治疗的情况下观察到的相比,在治疗期间和/或之后预防、最小化或减弱耐药性或难治性症状或病症。最小化、减弱或预防的难治性病症可以例如通过本领域技术人员所设想的临床评分来限制。在一个实例中,当本领域技术人员预期的评分低于在没有本文所述的治疗时观察到的评分时,应认为成功治疗了难治性或耐药性ms自身免疫性疾病。在一些方面,耐药性或难治性自身免疫性疾病可以是以下一种或多种:天疱疮、视神经脊髓炎/视神经脊髓炎-谱系障碍(nod/nmod)和多发性硬化症(ms),以及全身性疾病,如anca相关血管炎、类风湿性关节炎(ra)、克罗恩病、炎症性肠病(ibd)和系统性红斑狼疮(sle)。
[0155]
在一个特定方面,耐药性或难治性自身免疫性疾病是多发性硬化症(ms)。
[0156]
本发明提供治疗对小分子、肽制剂、蛋白质生物制剂、b细胞耗竭剂具有耐药性或难治性的自身免疫性疾病的方法。在一个优选的方面,本发明提供了治疗对b细胞耗竭剂具有耐药性或难治性的自身免疫性疾病的方法。在更优选的方面,本发明提供了治疗对上述常规疗法具有耐药性或难治性的ms的方法。在更优选的方面,本发明提供治疗对b细胞耗竭疗法具有耐药性或难治性的ms的方法。
[0157]
在本发明的替代方面,本发明的治疗方法包括将有效量的本文所述的化合物施用于患有自身免疫性疾病的哺乳动物。在另一个方面,本发明的治疗方法包括将有效量的本文所述的化合物施用于患有耐药性或难治性自身免疫性疾病的哺乳动物。在又一方面,本发明提供治疗对小分子、肽制剂、蛋白质生物制剂、b细胞耗竭剂与第二药剂的组合具有耐药性或难治性的自身免疫性疾病的方法。
[0158]
在又一方面,本发明的治疗包括将有效量的本文所述化合物单独施用或与第二小分子、肽制剂、蛋白质生物制剂和/或b细胞耗竭剂同时或依次组合施用。聚乙二醇化的t-bsab可以与另一种药剂同时施用或在另一种药剂施用之后施用。因此,用于本发明的化合物可以在第二药剂的治疗期间或之后施用。例如,第二药剂的非限制列表包括:小分子药剂
如tecfidera、芬戈莫德、tysabri、奥巴捷、克拉屈滨;肽制剂如克帕松、蛋白质生物制剂如ifn-β-1a、ifn-β-1b、抗cd52抗体(阿仑珠单抗,alemtuzumab)、那他珠单抗;b细胞耗竭剂如抗cd19药剂(伊奈利珠单抗、obexelimab)、抗cd20药剂(利妥昔单抗、奥瑞珠单抗、奥法木单抗)。
[0159]
在本发明的某些优选实施方案中,治疗自身免疫性疾病的方法包括施用具有下式的化合物:
[0160][0161]
其中mpeg的分子量选自10000至40000;a和b各自是独立地选自1至20的整数;x选自c、n、o;r1和r2各自独立地选自c
1-10
烷基或环烷基。
[0162]
在本发明的一个具体实施方案中,治疗自身免疫性疾病的方法包括施用具有以下结构的化合物:
[0163][0164]
其中,scacd19(抗cd19 scfv)具有以下氨基酸序列:
[0165][0166]
和
[0167]
scacd3(抗cd3 scfv)具有以下氨基酸序列:
[0168][0169]
具有上述结构的化合物在本文中称为“jy108”。
[0170]
k.术语定义
[0171]
如本文所用,术语“烷基”是指烃链,通常长度为约1至25个原子。这种烃链优选但不一定是饱和的并且可以是支链或直链,但通常直链是优选的。术语c
1-10
烷基包括具有1、
2、3、4、5、6、7、8、9和10个碳的烷基。同样地,c
1-25
烷基包括具有1至25个碳原子的所有烷基。示例性烷基包括甲基、乙基、异丙基、正丁基、正戊基、2-甲基-1-丁基、3-戊基、3-甲基-3-戊基等。如本文所用,当提及三个或更多个碳原子时,“烷基”包括环烷基。除非另有说明,否则烷基可以是取代的或未取代的。
[0172]
如本文所用,术语“官能团”或“官能团”是指在正常的有机合成条件下,可用于在其所附接的实体和通常携带其他官能团的另一实体之间形成共价连接基的基团。“双官能接头”是指具有两个官能团的接头,其通过与缀合物的其他部分形成两个连接基。
[0173]
如本文所用,术语“衍生物”是指具有附加结构部分的化学修饰化合物,目的是引入新的官能团或调整原始化合物的性质。
[0174]
如本文所用,术语“保护基团”是指在某些反应条件下防止或阻断分子中特定化学反应性官能团反应的部分。
[0175]
如本文所用,术语“peg”或“聚(乙二醇)”是指聚(环氧乙烷)。用于本发明的peg通常包含-(ch2ch2o)
n-的结构。peg可以具有多种分子量、结构或几何形状。peg基团可以包含在典型合成反应条件下不易发生化学转化的封端基团。封端基团的实例包括-oc
1-25
烷基或-o芳基。
[0176]
如本文所用,术语“接头”或“连接基”是指用于连接互连部分(如抗体和聚合物部分)的原子或原子集合。接头可以是可裂解的或不可裂解的。可裂解接头包含在某些生物或化学条件下可裂解的基团或部分。实例包括酶促裂解的二硫连接基、1,4-或1,6-苄基消除、三甲基锁系统、基于二(羟乙基)甘氨酸(bicine)的自裂解系统、酸不稳定的甲硅烷基醚接头和其他光不稳定的接头。
[0177]
如本文所用,术语“连接基团”或“连接基”是指连接化合物或缀合物的不同部分的官能团或部分。连接基团的实例包括但不限于:酰胺、酯、氨基甲酸酯、醚、硫醚、二硫化物、腙、肟和半碳二酰肼、碳二亚胺、酸不稳定基团、光不稳定基团、肽酶不稳定基团和酯酶不稳定基团。例如,接头部分和聚合物部分可以通过酰胺或氨基甲酸酯连接基团彼此连接。
[0178]
如本文所用,术语“肽”、“多肽”和“蛋白质”互换地用于描述聚合物中氨基酸残基的排列。肽、多肽或蛋白质可以由除了稀有氨基酸和合成氨基酸类似物外的标准的20种天然氨基酸组成。它们可以是氨基酸的任何链,无论何种长度或是否有翻译后修饰(例如,糖基化或磷酸化)。
[0179]“重组”肽、多肽或蛋白质是指通过重组dna技术产生的肽、多肽或蛋白质;即,由编码期望肽的外源dna构建体转化的细胞产生的肽、多肽或蛋白质。“合成的”肽、多肽或蛋白质是指通过化学合成制备的肽、多肽或蛋白质。当与参考例如细胞、或核酸、蛋白质或载体一起使用时,术语“重组”表示细胞、核酸、蛋白质或载体已通过引入异源核酸或蛋白质或改变天然核酸或蛋白质而被修饰,或细胞衍生自如此修饰的细胞。含有一种或多种前述序列和异源序列的融合蛋白在本发明范围内。异源多肽、核酸或基因源自外来物种,或者如果来自相同物种,则基本上从其原始形式修饰。如果两个融合的结构域或序列在天然存在的蛋白质或核酸中彼此不相邻,则它们彼此是异源的。
[0180]“分离的”肽、多肽或蛋白是指已经从与其天然结合的其它蛋白、脂质和核酸分开的肽、多肽或蛋白。多肽/蛋白质可以构成纯化制剂干重的至少10%(即,10%和100%之间的任何百分比,例如20%、30%、40%、50%、60%、70%、80%、85%、90%、95%和99%)。纯
度可以通过任何适当的标准方法测量,例如通过柱色谱法、聚丙烯酰胺凝胶电泳或hplc分析测量。本发明中描述的分离的多肽/蛋白可以从天然来源纯化、通过重组dna技术或通过化学方法产生。
[0181]“抗原”是指引发免疫反应或与该反应的产物结合的物质。术语“表位”是指抗体或t细胞结合的抗原区域。
[0182]
如本文所用,“抗体”以最广义使用,具体涵盖单克隆抗体(包括全长单克隆抗体)、多克隆抗体、多特异性抗体(例如,双特异性抗体)和抗体片段,只要它们表现出期望的生物活性即可。
[0183]
如本文所用,“抗体片段”可以包含完整抗体的一部分,通常包括完整抗体的抗原结合区和/或可变区和/或保留fcr结合能力的抗体的fc区。抗体片段的实例包括直链抗体;单链抗体分子;和由抗体片段形成的多特异性抗体。优选地,抗体片段保留igg重链的整个恒定区,并且包括igg轻链。
[0184]
如本文所用,术语“fc片段”或“fc区”或“fc”用于限定免疫球蛋白重链的c端区域。
[0185]
本文使用的术语“传统抗体”是指全长单克隆抗体或其修饰形式。
[0186]
如本文所用,术语“单克隆抗体”是指从一群基本同源抗体获得的抗体,即构成该群体的单个抗体除了可能少量存在的可天然发生的突变以外是相同的。单克隆抗体是高度特异性的,针对单一的抗原位点。此外,与通常包括针对不同决定簇(表位)的不同抗体的常规(多克隆)抗体制剂相反,各单克隆抗体针对抗原上的单个决定簇。修饰语“单克隆”表明从基本同源的抗体群体获得的抗体的特征,并不理解为需要通过任何特定的方法来产生该抗体。例如,根据本发明使用的单克隆抗体可以通过kohler和milstein,1975,nature,256,p495-497首先描述的杂交瘤方法制备,其通过引用并入本文,或者可以通过重组dna方法制备(参见例如,美国专利号4,816,567,其通过引用并入本文)。单克隆抗体也可以使用clackson等人,1991,nature,352,p624-628和marks等人,1991,j mol biol,222,p581-597中描述的技术从噬菌体抗体文库中分离,其中每篇都通过引用并入本文。
[0187]
本文中的单克隆抗体具体包括“嵌合”抗体(免疫球蛋白),其中重链和/或轻链的一部分与衍生自特定物种或属于特定抗体类别或亚类的抗体中的相应序列相同或同源,而链的其余部分与衍生自另一物种或属于另一抗体类别或亚类的抗体以及此类抗体的片段中的相应序列相同或同源,只要它们表现出期望的生物活性即可(参见美国专利号4,816,567;morrison等人,1984,proc natl acad sci usa,81,p6851-6855;neuberger等人,1984,nature,312,p604-608;takeda等人,1985,nature,314,p452-454;国际专利申请号pct/gb85/00392,其中每篇都通过引用并入本文)。
[0188]“人源化”形式的非人(例如鼠)抗体是嵌合抗体,其含有衍生自非人免疫球蛋白的最小序列。对于绝大部分而言,人源化抗体是人免疫球蛋白(受体抗体),其中来自受体的超变区的残基被来自非人物种(供体抗体)的具有期望的特异性、亲和力和能力的超变区的残基替代,所述非人物种如小鼠、大鼠、兔或非人灵长类动物。在某些情况下,人免疫球蛋白的fv框架区(fr)残基被相应的非人残基所替代。此外,人源化抗体可以包含未在受体抗体或供体抗体中出现的残基。进行这些修饰来进一步优化抗体的性能。通常,人源化抗体包含至少一个且通常包含两个可变结构域的基本全部,其中所有或基本所有的超变环对应于非人免疫球蛋白的那些超变环,并且所有或基本所有的fr残基是人免疫球蛋白序列的那些残
基。任选地,人源化抗体还包含免疫球蛋白恒定区(fc)的至少一部分,通常是人免疫球蛋白的恒定区(fc)的至少一部分。更多细节参见jones等人,1986,nature,321,p522-525(1986);riechmann等人,1988,nature,332,p323-327;presta,2003,curr op struct biol,13(4),p519-525;美国专利号5,225,539,其中每篇都通过引用并入本文。“人抗体”是指具有完全人序列的任何抗体,如可以从人杂交瘤、人噬菌体展示文库或表达人抗体序列的转基因小鼠获得的抗体。
[0189]
术语“药物组合物”是指活性剂与惰性或活性载剂的组合,使得该组合物特别适用于体内或离体的诊断或治疗用途。
[0190]
如本文所用,“药学上可接受的载剂”包括生理上相容的任何和所有溶剂、分散介质、包衣、抗细菌剂和抗真菌剂、等渗剂和吸收延迟剂等。“药学上可接受的载剂”在向受试者施用或施用于受试者后不会引起不期望的生理作用。药物组合物中的载剂也应该是“可接受的”,因为其与活性成分相容并且可以能够稳定活性成分。一种或多种增溶剂可用作药物载剂以递送活性剂。药学上可接受的载剂的实例包括但不限于生物相容性载体、佐剂、添加剂和稀释剂,以获得可用作剂型的组合物。其他载剂的实例包括胶体氧化硅、硬脂酸镁、纤维素和十二烷基硫酸钠。
[0191]
其他合适的药物载剂和稀释剂,以及它们使用的药物必需品,描述于remington's pharmaceutical sciences中。优选地,载剂适用于静脉内、肌肉内、皮下、肠胃外、脊柱或表皮施用(例如,通过注射或输注)。治疗化合物可以包括一种或多种药学上可接受的盐。“药学上可接受的盐”是指保留母体化合物的期望生物活性并且不赋予任何不期望的毒理学作用的盐(参见例如,berge,s.m.,等人1977,j.pharm.sci.66,p1-19)。
[0192]
如本文所用,“治疗”或“疗法”是指向患有病症或有发展病症风险的受试者施用化合物或药剂,目的是治愈、缓解、减轻、补救、延缓其发作、预防或改善病症、病症的症状、继发于病症的疾病状态或对病症的易感性。
[0193]“有效量”是指赋予治疗对象治疗效果所需的活性化合物/药剂的量。如本领域技术人员所认识到的,有效剂量将根据所治疗病症的类型、施用途径、赋形剂的使用以及与其他治疗性治疗共同使用的可能性而变化。用于治疗自身免疫性疾病的组合的治疗有效量是与未治疗的受试者相比将导致例如临床评分降低或减缓症状进展的量。
[0194]
如本文所公开的,提供了一些数值的范围。应当理解,除非上下文另有明确规定,否则还具体公开了该范围的上限和下限之间的每个中间值,至下限单位的十分之一。在所述范围内的任何规定值或中间值与所述范围内的任何其他陈述或中间值之间的每个较小范围都涵盖在本发明内。这些较小范围的上限和下限可以独立地包括在该范围内或排除在该范围外,并且在较小范围内包括任一个、零个或两个限制的每个范围也涵盖在本发明内,受制于所述范围内任何特别排除的限制。在所述范围包括一个或两个限制的情况下,排除那些包括的限制之一或两者的范围也包括在本发明中。
[0195]
术语“约”通常是指所示数字加或减10%。例如,“约10%”可表示9%至11%的范围,而“约1”可表示0.9-1.1。“约”的其他含义可以从上下文中显而易见,如四舍五入,在这种情况下,例如“约1”也可以表示从0.5至1.4。
实施例
[0196]
以下实施例用于提供对本发明的进一步理解,但并不意味着以任何方式限制本发明的有效范围。
[0197]
实施例1:聚乙二醇化bsab抗cd3x抗cd19(jy108)的靶标结合
[0198]
由于对tcr具有强亲和力的t-bsab(靶向cd3和肿瘤相关抗原)如博纳吐单抗与通常导致临床上严重的crs的过度的细胞因子释放相关,因此通过biacore测定和elisa测定评估了jy108的靶标结合。
[0199]
1)biacore测定
[0200]
设备:biacore
[0201]
样品:抗cd3 scfv(scacd3)、抗cd3 scfv-peg(抗cd3-peg)、抗cd19 scfv(scacd19)、抗cd19 scfv-接头(抗cd19-接头)、jy108和博纳吐单抗。抗cd3 scfv(scacd3)具有seq id no:2所示的氨基酸序列。抗cd19 scfv(scacd19)具有seq id no:1所示的氨基酸序列。wo 2018/075308 a1中详细描述了这些样品的制备方法。
[0202]
一般程序:稀释的人cd19(acrobiosystems,cat.cd19-h5259)或人cd3e&3d(acrobiosystems,cat.cdd-h52w0)通过fc捕获方法捕获在传感器芯片上。人cd3e&cd3d和人cd19的捕获时间分别设置为12s和12-30s,捕获流速为10l/min。每个测试的样品都被连续稀释成6-8个浓度。分析物的结合时间和解离时间分别设置为180s和420s,分析物的流速设置为30l/min。对于表面再生,使用10mm glycine-hcl缓冲液;接触时间设置为30s,流速为30l/min。每个循环中流动池1和缓冲液的空白进样用作响应单位(response units)减法的双重参考。所有数据均使用biacore t2000评估软件(3.1版)进行处理。结合动力学数据如表1所示。
[0203]
如表1所示,jy108对人cd3e&cd3d的kd(结合亲和力)为3.31
×
10-8
m,而博纳吐单抗的kd为1.14
×
10-9
m。jy108的亲和力比博纳吐单抗弱30倍左右。jy108对cd3的亲和力降低可能是由大peg的阻碍作用引起的,因为抗cd3 scfv-peg对靶标的亲和力也比非聚乙二醇化的抗cd3 scfv弱(14倍)。
[0204]
类似地,jy108对人cd19的kd为1.44
×
10-9
m,而博纳吐单抗的kd为2.07
×
10-10
m。同样,由peg引起的阻碍效应可能导致jy108对人cd19的亲和力减弱。正如预期的那样,针对人cd19的抗cd19 scfv接头(接头是小得多的peg
11
)的kd为1.11
×
10-10
m,与抗cd19 scfv的kd(6.53
×
10-11
m)相似。
[0205]
表1.抗体对人cd19/人cd3e&3d的亲和力测量
[0206][0207]
2)elisa测定
[0208]
为了评估与人cd3e&cd3d蛋白的结合,用cbs制备scacd3、cd3-peg(聚乙二醇化scacd3)、博纳吐单抗和3批jy108(分别为ph 6.8缓冲液中2批和ph 4.7缓冲液中1批)的样品溶液,每个样品的四种浓度为30、1和0.01μg/ml。板分别以100μl/孔包被有scacd3、cd3-peg和jy108。类似地,为了评估与人cd19蛋白的结合,用cbs制备scacd19、cd19-接头、博纳吐单抗和三批jy108(分别为ph 6.8缓冲液中2批和ph 4.7缓冲液中1批)的样品溶液,每个样品的两种浓度为10、1μg/ml,并以100μl/孔包被到板上。包被后,每块板用pbst(200μl/孔)洗涤3次。接下来,将每个板用pbst中的5% bsa在37℃封闭2小时。封闭后,每个板用pbst洗涤3次,然后加入1μg、0.1μg和0μg于0.5% bsa于pbst中的人cd19-fc蛋白(acrobiosystems,代码:cd9-h5259)(10μg/ml、1μg/ml、0μg/ml,100μl/孔)或1μg 0.5% bsa于pbst中的人cd3e&cd3d(acrobiosytems,代码:cdd-h52w0)(10μg/ml、100μl/孔)到相应的孔中。将板在37℃下孵育1小时,然后用pbst洗涤3次。然后将100μl的1μg/ml hrp缀合的抗人igg(genscript,代码:a01854)添加到每个孔中。将板在37℃孵育1.5小时。孵育后,将每个板用pbst洗涤3次,然后向每个孔中加入100μl tmb。每个板的显影反应设置为在黑暗中15分钟,并以100μl/每孔的终止溶液终止。用酶标仪在od450 nm处读取每个板,结果如图1所示。
[0209]
图1中的结果表明,在1μg/ml的包被浓度下,测试样品对人cd3e&cd3d的亲和力可排列为:scacd3》博纳吐单抗》jy108》scacd3-peg。还观察到与人cd19结合的样品的类似排名(scacd19》博纳吐单抗》jy108》scacd19-接头)。
[0210]
结论:biacore测定和elisa测定的结果一致,表明聚乙二醇化t-bsab jy108对cd3和cd19的亲和力显著降低。在这两种测定中观察到的通过聚乙二醇化降低的亲和力有望为jy108带来有益的效果,例如较少的细胞因子释放。
[0211]
实施例2:jy108对cd19
+
细胞系和正常b细胞的体外功效
[0212]
尽管与靶标的结合减少,但为了测试聚乙二醇化t-bsab jy108对多种靶细胞的功效,进行了以下实验。
[0213]
一般程序:在100μl培养基中以4
×
104、10
×
104或20
×
104(e:t比率分别为2:1、5:1或10:1)的扩增效应细胞与指定剂量的jy108一起室温下孵育半小时。加入2
×
104个cd19
+
或cd19-靶细胞(raji、nalm-6、reh或k562),并在37℃下孵育24小时或根据指示孵育。对于仅
含效应细胞的孔,添加100μl不含靶细胞的培养基,对于仅含靶细胞的孔,添加100μl不含效应细胞的培养基。每孔加入20μl mts试剂,按照制造商手册在37℃下孵育。
[0214]
孵育后,记录od
490
nm处的吸光度,根据公式计算死细胞百分比:
[0215]
细胞毒性%=1-(od
实验-od
pbmc
)/(od
靶标-od
培养基
)
[0216]
其中od
实验
是指含有jy108、效应细胞和靶标的孔在设计的e:t比率下的od
490
。od
pbmc
是指具有指定jy108剂量的仅效应细胞而没有靶细胞的od
490
。od
靶标
是指仅具有靶细胞而没有jy108也没有效应细胞的od
490
。od培养基是指等体积培养基而不含jy108、效应细胞或靶细胞的od
490
。该程序用于测定jy108对除pbmc中的b细胞之外的所有细胞系的体外功效,在这种情况下,使用荧光抗体染色,然后进行流式细胞术检测。
[0217]
1)jy108对cd19-细胞无药物特异性细胞毒性
[0218]
为了测试jy108是否靶向cd19-细胞,本实验使用了具有cd19阴性表达的白血病细胞系k562。图2的结果表明,即使在e:t=5:1的1μg/ml浓度下,存在或不存在jy108的细胞杀伤效果没有显著差异(阴性对照无jy108)。结果表明jy108是安全的,不会对cd19阴性细胞产生药物特异性细胞毒性。
[0219]
2)jy108从健康供体中消除pbmc中的b细胞
[0220]
为了测试jy108是否能消除pbmc中的cd19阳性细胞,将4种不同浓度的jy108(对照0μg/ml、0.1μg/ml、1μg/ml和10μg/ml)加入pbmc中,37℃孵育24小时。孵育后,pbmc样品用1μl fitc标记的抗cd19染色30分钟,然后用流式细胞术检测。结果显示于图3中。与对照样品(不含jy108)中6.93%的cd19阳性细胞相比,jy108在0.1μg/ml浓度下显著降低cd19阳性细胞至背景水平。这一结果清楚地表明,jy108可以有效地消除pbmc中的cd19阳性细胞。
[0221]
3)e:t比率对jy108功效的影响
[0222]
在不同的e:t比率下,jy108对相同的靶细胞表现出不同的毒性。图4中的结果显示,jy108在e:t=5:1时对nalm-6杀伤的ec
50
为37.56ng/ml,而jy108在e:t=10:1时的ec
50
为14.68ng/ml。结果表明,效应细胞越多,实现期望的细胞杀伤效果所需的jy108就越少。然而,当其他参数保持不变时,jy108的细胞毒性随着e:t比率的增加而增加的状况达到稳定水平(数据未显示)。
[0223]
4)细胞系对jy108功效的影响
[0224]
jy108对不同的cd19
+
细胞系有不同的杀伤作用(图5)。在实验环境下,jy108针对reh的ec
50
为10.38ng/ml,而jy108针对raji细胞的ec
50
为159.3ng/ml。
[0225]
结论:jy108的ec
50
在具有不同的e:t比率、不同的pbmc供体、不同的作用持续时间和不同的cd19阳性细胞系的所有测定中均处于ng/ml水平,范围从个位数ng/ml到数百ng/ml。病理性cd19
+
细胞的体外杀伤具有药物特异性,符合jy108药物设计的预期。
[0226]
实施例3:jy108对前-b all肿瘤模型的体内功效
[0227]
为了测试jy108的体内功效,收集并计数reh肿瘤细胞,然后将细胞浓度调整至10
×
107/ml。将扩增的pbmc细胞调整为30
×
107/ml。在接种到动物之前,将等体积的扩增bmc细胞和reh肿瘤细胞混合。jy108用无菌pbs制备至浓度分别为200μg/ml、50μg/ml和12.5μg/ml。小鼠随机分为5只/组。对制备的jy108进行尾静脉施用(100μl/小鼠)。注射jy108后,将等体积混合的reh和扩增的pbmc细胞皮下接种到小鼠体内(200μl/小鼠)。
[0228]
图6的结果表明,jy108在1.25μg的剂量下可以有效抑制reh肿瘤的生长。实验组小
鼠与对照组小鼠体重无显著差异,表明实验剂量的jy108对动物没有产生毒性作用。
[0229]
实施例4:jy108的药代动力学特性
[0230]
jy108包含his标记的scacd3和不带它的标记的scacd19。因此,抗his标签抗体和cd19抗原可用在elisa中用于检测完整的jy108分子。在实施例1中描述的类似elisa测定中,人cd19-fc以1μg/孔用作包被剂,抗his用作检测抗体。图7中显示的结果表明用于野生型c57bl/6中的jy108的t
1/2
为24.28小时。
[0231]
jy108在受试动物中的半衰期比对照药物博纳吐单抗的半衰期(据报道为约2小时)长得多。由于啮齿类动物的聚乙二醇化蛋白的代谢比人体快5倍左右(us 2011/0112021 a1),因此jy108在人体内的半衰期预计将大于120小时,这将大大有利于jy108的临床疗效。
[0232]
实施例5:选择人源化转基因小鼠(hcd3ehcd19)作为jy108毒性评价的动物模型
[0233]
在这项研究中,构建了针对两种靶基因(cd3ε和cd19)的人源化转基因小鼠,并用于评估该分子的毒性和体内功效。
[0234]
1)jy108与动物模型靶标的结合(图8)
[0235]
scacd3、scacd19和jy108分别根据供应商的方案进行了fitc标记。收集hcd3ehcd19转基因小鼠的脾组织,在淋巴细胞分离液中研磨,然后离心获得悬浮的小鼠淋巴细胞。收集并洗涤淋巴细胞后,使用冷的facs缓冲液(含3% fbs的pbs)将细胞悬液调节至5
×
106个细胞/ml。将重悬的细胞以100μl/管等分。将fict(或pe、或apc)标记的抗体添加到细胞混悬液中,并将反应混合物在4℃的黑暗中孵育30分钟。随后,通过在每次洗涤时以400g离心5分钟,用facs缓冲液洗涤细胞3次。在使用流式细胞术分析细胞之前,使用500μl facs缓冲液(补充3% fbs的冷pbs)重悬细胞。
[0236]
在图8a中,流式细胞术结果显示重组人cd3(fitc-hcd3)在鼠t细胞(pe-mtcr-b)上表达,重组人cd19(apc-hcd19)在鼠b细胞(pe-mcd45)上表达。在图8b中,结果表明来自转基因小鼠的10.7%脾细胞由鼠tcr-b和fitc标记的scacd3双重染色,53.6%的来自转基因小鼠的脾细胞对鼠tcr-b和fitc标记的jy108呈双阳性。在图8c中,33.3%的来自转基因小鼠的脾细胞对鼠cd45r和scacd19呈双阳性,13.1%的来自转基因小鼠的脾细胞对鼠cd45r和jy108呈双阳性。图8a、8b和8c的综合结果表明,来自hcd3ehcd19转基因小鼠的淋巴细胞被scacd3、scacd19和jy108特异性结合。
[0237]
2)来自转基因小鼠(hcd3hcd19)的脾细胞作为jy108的效应细胞
[0238]
对来自转基因小鼠的t细胞进行了评估,发现这些t细胞可用作jy108体外细胞毒性测定中预期的效应细胞。与实施例2的结果相似,hcd3ehcd19转基因小鼠的脾细胞也能有效杀伤raji、nalm-6和reh靶细胞,如图9所示。
[0239]
结论:以上结果表明转基因小鼠hcd3hcd19是可用于jy108毒性评价的动物模型。
[0240]
实施例6:10mg/kg的单剂量jy108无急性毒性
[0241]
以10mg/kg将jy-108静脉内给药hcd3ehcd19转基因小鼠(n=3,单剂量)用于急性毒性观察。与阴性对照组相比,实验组hcd3ehcd19纯合转基因小鼠的体重(p》0.05)、摄食量无显著变化,但耗水量略有增加(图10)。
[0242]
实施例7:jy108导致从人全血中释放更少的细胞因子
[0243]
细胞因子风暴或细胞因子释放综合征是t细胞衔接抗体药物(例如,博纳吐单抗)的主要临床副作用之一。为了评估与对照药物博纳吐单抗相比,jy108亲和力降低是否会导
致更少的细胞因子释放和提升的安全性,jy108和博纳吐单抗均在不同浓度(0.0005μg/ml、0.005μg/ml、0.05μg/ml、0.5μg/ml和5μg/ml)下与来自2个健康供体的人全血(hwb)一起孵育24小时。使用bd
tm
cytometric bead array(cba)人th1/th2(目录号:551809)通过流动微球技术测量孵育2小时、6小时和24小时时所释放的细胞因子。计算了两个供体的平均值。每种细胞因子在24小时内的最高值如图11所示。结果表明,jy108诱导的细胞因子释放包括il2、il6、tnf、ifn-γ和il10,它们均低于对照药物博纳吐单抗诱导的相应释放。因此,可以预测,与博纳吐单抗相比,jy108在体内引起的细胞因子风暴和相关副作用显著降低。
[0244]
实施例8:jy108显著改善eae
[0245]
本研究的目的是测试jy108对实验性自身免疫性脑脊髓炎(eae)(用于ms的自身免疫性疾病模型)的功效。
[0246]
1)eae模型的生成
[0247]
按照标准方案在c57bl/6转基因小鼠(hcd3ehcd19纯合子)上生成eae模型。在生成模型之前,在pbs中制备100μg/ml百日咳毒素(ptx)储备溶液并保持在4℃。在每次注射当天,将ptx储备溶液在pbs中稀释至1μg/ml的工作溶液。在第0天,等体积的mog
1-125
蛋白(2mg/ml)和完全弗氏佐剂(cfa,4mg/ml)在均质器中完全乳化,直到混合物变得均匀。然后将mog
1-125
和cfa的乳剂在小鼠胸腹部两侧皮下注射(150μl/小鼠),随后以200μl/小鼠尾静脉注射ptx。在第2天,以200μl/小鼠进行第二次尾静脉注射ptx。每天测量每只小鼠的体重,并根据kono的5分标准(1级:尾巴不挺直;2级:尾巴不挺直且后肢无力;3级:部分后肢瘫痪;4级:完全后肢瘫痪;5级:垂死状态)(hou,y.等人2013,stem cell res ther 4,77)用临床评分(gs)对动物的疾病症状进行评估。
[0248]
2)jy108显著减轻eae症状
[0249]
eae症状在第11天开始出现在转基因小鼠中,并且随着时间的推移继续恶化。在第16天,cs达到3。然后在第16天将有症状的eae模型小鼠随机分组,以便实验组和对照组具有相似的临床评分和体重(每组n=4)。实验组以60μg/动物隔日一次尾静脉施用jy108(共7次),第28天停药,对照组(图12中模型组)静脉注射载体。监测两组小鼠的疾病症状,并每天记录其cs。
[0250]
如图12所示,jy108组在初次施用后症状明显缓解,后期出现反弹,之后稳定在相对较低的cs。未添加jy108的模型组(对照组)小鼠在分组开始时显示症状的加重趋势,随后略有缓解,但症状评分保持在相对较高水平。体重曲线似乎与eae的临床症状特征密切相关:病症严重时体重低,病症缓解时体重高。eae病症可能会影响小鼠的活动能力,包括获取食物的能力。
[0251]
3)组织病理学切片的lfb染色
[0252]
为了进一步支持观察到的改善eae症状的证据,根据病理学测试程序对小鼠的脊髓和脑组织进行染色。使用nikon(eclipse ci-l)显微镜和带有caseviewer2.2的3dhistech切片扫描仪(pannoramic desk/midi/250/1000)来获取图像。
[0253]
lfb(luxol fast blue)染料可被髓鞘脂蛋白吸引,对cns组织进行特异性染色。结果显示于图13中。对于脊髓,lfb染色结果显示健康动物白质髓鞘一致,可均匀染色,未见明显脱髓鞘。在模型组中,白质周围大面积消失,lfb染色的髓鞘变浅且不连续,表明多处神经纤维脱髓鞘。相比之下,jy108处理动物的脊髓显示出更多的lfb染色区域,并且染色比eae
模型组更连续。结果表明,jy108大大降低了模型中脱髓鞘的严重程度。
[0254]
基于临床症状改善、体重变化和组织lfb染色病理切片分析结果,可以得出jy108有效改善eae症状的结论。
[0255]
4)jy108耗竭mog特异性自身抗体
[0256]
为了进一步了解jy108在eae小鼠中的功效,在第22天检查了来自动物的mog特异性自身抗体。除了用mog
1-125
蛋白包被板之外,与实施例1和4中描述的elisa方法类似的程序用于该测定。图14中显示的结果表明jy108有效降低了血清中mog特异性抗体水平(图14)。
[0257]
实施例9:jy108与medi-551的头对头比较
[0258]
medi-551(伊奈利珠单抗)是抗cd19单克隆抗体,目前正在临床试验中用于治疗ms。临床前结果表明medi-551在eae模型中比上市的单克隆抗cd20抗体更有效。对于头对头比较,本实验中的eae模型以与实施例8中所述相同的方式构建,并且在实验中包括medi-551处理组(单次静脉注射250μg/动物)。在eae模型生成后第12天,将雄性动物随机分为4组(n=7):medi-551处理组、模型组(对照组)、用总共5剂(第14天、第15天、第17天、第19天和第21天)的jy108以60μg/动物和80μg/动物处理的两组。每天记录每只动物的临床评分(第18天除外)。雌性小鼠在eae诱导第11天后进行类似分组(n=7),并用jy108(30、50、75μg/动物)和medi551(250μg/动物)处理。
[0259]
如图15所示,与赋形剂处理的eae模型组相比,以60μg/动物和80μg/动物给药的jy108显著改善了雄性小鼠的eae症状。此外,jy108在80μg/动物时比60μg/动物时更有效地改善雄性小鼠的eae症状。用30、50和75μg jy108/动物处理的雌性eae小鼠也获得了类似的结果,而用medi551的处理无效。尽管据报道在eae诱导后第7天施用medi-551能够延迟雄性小鼠的eae症状(chen,d.等人2014,j immunol 193,4823-4832),但令人惊讶的是,相同的化合物在本实验第14天施用时对严重患病的eae动物没有显示出任何治疗效果(p》0.05)(图15)。
[0260]
实施例10:jy108在改善eae中的机制
[0261]
1)jy108部分去除cd19
+
b细胞
[0262]
为了更好地说明jy108改善eae症状的机制,在第15天用实施例9中描述的相同方法检查eae实验中的脾b细胞。结果显示于图16中。令人惊讶的是,虽然medi-551几乎完全消除了cd19
+
b细胞,但jy108仅部分去除了cd19
+
b细胞:从eae模型中的68.2%到jy108处理组的37.1%(60μg/动物)和39.1%(80μg/动物)。
[0263]
2)b
reg
细胞对medi-551和jy108均具有耐药性
[0264]
jy108在改善eae症状方面表现出比medi-551更高的功效(图15),但在耗竭总cd19
+
b细胞方面不如medi-551有效(图16)。看似矛盾的结果促使了解jy108处理后其他相关免疫细胞发生了什么。使用实施例5中描述的方法,衍生自eae实验中的动物的脾细胞用pe-抗cd19和fitc-b10(breg的标志物)双重染色并通过流式细胞术计数。如图17a所示,所有五组中免疫抑制性breg细胞的水平相似,这表明jy108和medi551在收集样品时对breg细胞的影响很小。
[0265]
由于mog特异性b细胞是cns髓鞘损伤自身抗体的主要来源,因此按照供应商手册提供的方案进行elispot,以比较受试动物脊髓中残留的mog特异性b细胞。图17b所示的结果与图16中的结果一致,表明jy108和medi551均有效地耗竭了mog特异性b细胞。
[0266]
结论:图17的结果表明可能涉及其他机制,导致jy108在改善eae症状方面比medi551具有更高的功效。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1