一种海洋链霉菌S187中小蛋白预测和鉴定方法
本发明涉及细菌中微肽的鉴定,具体而言,涉及一种海洋链霉菌s187中小蛋白预测和鉴定方法。
背景技术:
1、小开放阅读框(small open reading frames,smorfs)是真核生物和细菌中可以从少于100个密码子翻译出来的dna序列。它们广泛分布在各种物种的基因组中,并且越来越多的研究表明它们具有重要功能。然而,smorf通常根据其小大或者特殊结构被认为是非编码的而常常被人们忽视。
2、近年来,随着新一代测序(ngs)技术的进步,人们开发了各种基于生物信息学的方法来探索数以千计的smorf。质谱(ms)是直接测定和定量编码多肽(smorf encodedpolypeptides,seps)最通用的工具。然而质谱法鉴定seps仍然存在一些缺点如由于seps通常都表达量较低且分子量都比较小,在质谱的检测的过程中高表达量的大蛋白的肽段信号干扰而造成seps的漏检,因此需要针对于seps的特异性富集与高精密的检测方法以提高seps的鉴定准确性与全面性。相对于依赖数据采集(data-dependent acquisition,dda),数据独立采集(data-independent acquisition,dia)是蛋白质组学中的新兴技术,需要通过dda实验生成光谱库,相比之下,dia具有更好的数据利用率和可重复性。值得一提的是,dia可以检测不同生长期seps表达的动态变化,其方差系数低,肽定量精度高。此外主流的蛋白质谱研究技术“鸟枪法”是通过在复杂的蛋白质样品中用酶(常见的为胰蛋白酶)酶解成的肽混合物进行质谱检测,但酶切后的肽段会损失蛋白的信息或降低蛋白检测覆盖率。而小蛋白本身长度较小,因此自上而下的蛋白研究技术越来越受到学者们的青睐。
3、目前,数据库检索的鉴定方式因其精度高、操作简单而成为肽组学研究的主要方法。完全注释的蛋白质数据库已经可以用于人类、小鼠和常见的实验室模型生物。然而,由于缺乏可用的公共数据库,很难在非模式生物中识别新的seps。从头测序通过比较质谱与氨基酸残基的质量差异直接推断肽序列,避免了在发现更多新的seps时对数据库的依赖。从头测序还可以提高肽序列覆盖率,实现相当高的计算速度,并检测突变氨基酸。但是,它不能像数据库搜索那样提供关于表达式或注释seps功能的定量数据。
技术实现思路
1、为解决上述缺陷,本发明提供了一种海洋链霉菌s187中小蛋白预测和鉴定方法,本发明有助于在多肽测序中测到氨基酸长度更长的多肽和提高蛋白对应多肽的覆盖率,为后续分析提供更可靠的肽段定位和定量信息。
2、一种海洋链霉菌s187中小蛋白预测和鉴定方法,其包括以下步骤:
3、1)全面数据库的建立:对于非模式菌株定制包含基因组预测与转录组检测的数据库;
4、2)样品处理:提取不同代谢时间点的样品中的多肽并进行富集,然后将富集到的微肽进行高精密度dia质谱检测;
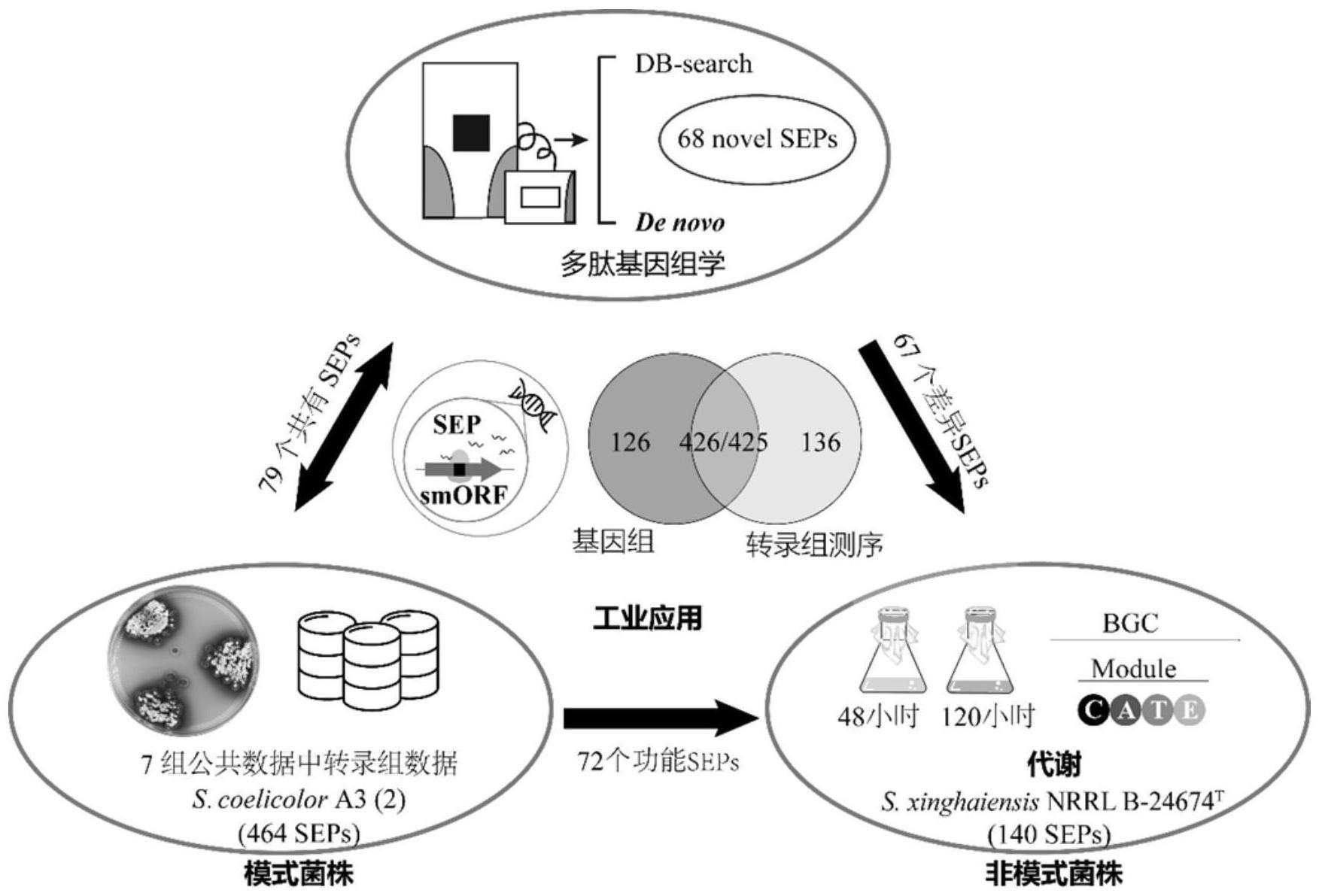
5、3)sep鉴定结果分析:通过质谱数据分析得到样品微肽的定性与定量结果,并将质谱数据同时采用数据库搜索与不依赖于数据库的de novo测序的鉴定方法寻找新颖的多肽与ncrna。
6、于本发明的一种实施方式中,步骤1)包括以下过程:
7、1.1)全基因组预测:选取两种软件对s187全基因组orf进行预测:
8、将s187全基因组fasta序列提交到orffinder进行六帧翻译,使用atg、gtg、ctg、ttg四种起始密码子,并选择代表细菌的11号密码子翻译表进行翻译;使用prodigal软件进行全基因组orf预测作为orffinder工具的补充,并由此得到orf对应的核糖体结合基序;运行参数选择代表细菌的11号密码子表,得到s187全基因组编码序列;对这些氨基酸序列长度进行统计,并筛选长度为100aa以下的orfs作为smorfs;
9、1.2)共线性分析:选取链霉菌属模式菌株天蓝色链霉菌作为参考,使用tbtools软件中的mcscanx工具与s187进行了基因组共线性分析以探究两菌株的相似关系;
10、1.3)培养、取样:
11、将星海链霉菌s187保藏菌株划线活化于tsb固体培养基,置于恒温培养箱,将活化后的菌种制备发酵种子液;
12、取样:先进行s187的发酵曲线和抗补体活性曲线等测定菌株活动周期,根据发酵曲线和抗补体活性曲线确定取样时间点:为36h、48h、72h、120h,然后取样、洗脱、保藏;
13、1.4)转录组测序建库:
14、将4个时间点的样品合并进行rna-seq建库,具体步骤如下:样品经过rna抽提、纯化、建库之后,采用ngs测序技术基于illumina hiseq测序平台,对这些文库进行paired双末端测序;对转录组测序得到的全部的转录本使用ncbi orffinder进行开放阅读框搜索,最小orf长度设置为30,genetic code设置为11.bacterial,orf起始密码子选择为任意起始密码子;将得到的转录组六帧翻译数据作为搜库数据库,并在多肽组测序流程中进行搜库;
15、转录组数据smrnas预测:对转录组测序数据使用fastaqc、trimmomatic进行质控,使用bowtie2和featurecounts分别进行reads比对定量,并对fpkm>1且length≤303nt的rna进行筛选,作为候选的smrnas库。
16、于本发明的一种实施方式中,在步骤1.3)中,将活化后的菌种制备发酵种子液包括以下步骤:
17、种子液接取:取tsb固体培养基活化好的s187菌块,加至tsb液体培养基,在210-230rpm震荡培养,36h后取样接取发酵液;
18、发酵液接取:取种子液加至m33培养基,在170-190rpm震荡培养,每48h转速增加20rpm,直至加至210-230rpm后转速维持不变。
19、于本发明的一种实施方式中,在步骤1.3)中,取样、洗脱、保藏包括以下步骤:
20、1.3.1)从同一批次的六瓶发酵液中取三瓶长势良好的取样,每瓶取样3ml加到同一50ml离心管中并混匀,剩余三瓶发酵液中其中一瓶取样9ml至50ml离心管,后续处理与第一瓶一致以用于配平,其余两瓶均取样10ml至离心管以待萃取旋蒸后检测产物,整个实验流程中离心管应一直放置于冰浴中;
21、1.3.2)去除培养基中蛋白质:离心,并弃去上清;
22、1.3.3)pbs洗脱:向离心沉淀中加入预冷的pbs溶液混合均匀,然后加入pbs涡旋振荡,离心并弃去上清;
23、1.3.4)重复步骤1.3.3)一次;
24、1.3.5)去除caco3:加入预冷的pbs溶液混合均匀,然后加入pbs涡旋振荡,离心并弃去沉淀,最后转移样品至新的离心管中;
25、1.3.6)再重复步骤1.3.3)一次,进行洗脱;
26、1.3.7)离心管吊入液氮罐中速冻,低温保存。
27、于本发明的一种实施方式中,步骤2)包括以下过程:
28、2.1)微肽的富集与处理:
29、小蛋白多肽提取:按发酵时间取样分离得到的s187菌丝体样本破碎,然后用甲醇:氯仿:水=3:1:4(v:v)提取小肽;涡旋,离心;收集上层水性上清液,用截留分子量为10kd的mwco在离心力在20000×g的条件下离心并取下层溶液,真空浓缩,干燥;脱盐冻干;
30、hplc分馏分:每个样本取一部分肽段混合,然后通过高效液相色谱,将肽段分离成多个馏分;
31、2.2)多肽组测序:
32、dda质谱上机:配制流动相a相和b相;使用10μl a相溶解冻干粉末,4℃下14000g离心20min,取上清1μg样品进样,液质连用检测;
33、dia质谱上机和搜库:使用0.1%(v/v)甲酸水溶液溶解多肽干粉并加入irt标准肽段,取1μg样品进样,进行液质联用检测。
34、于本发明的一种实施方式中,步骤3)包括以下步骤:
35、3.1)数据库搜索鉴定seps:使用t检验对样品多肽的定量值进行差异分析,卡pvalue≤0.05,foldchange≥1.5,得到差异肽段,再根据肽段差异情况使用投票法对其对应的蛋白的上下调情况进行统计,并筛选氨基酸序列长度在100aa以下的小蛋白作为所鉴定到的seps。
36、于本发明的一种实施方式中,步骤3)还包括以下步骤:
37、3.2)de novo测序重分析:根据多肽组测序得到的质谱数据,使用de novosequencing对质谱数据进行了重分析,以检测到数据库中可能不包含的novel seps,测序结果和从头测序中novel seps筛选过程如下:
38、3.2.1)de novo测序主要参数设置:
39、使用peaks studio多肽组测序质谱数据进行重分析,使用de novo sequencing的方法根据质谱分子量对肽段进行序列鉴定,并使用peaks db对鉴定到的肽段进行数据库搜库以确定肽段-蛋白归属,将de novo测序结果与数据库搜索结果结合以提供翻译后修饰和突变等全新肽段的附加信息,数据库搜索不到的肽段即被视为de novo only peptide;数据库设置为s187转录组测序所生成的转录组六帧翻译库,参数设置none enzyme即非酶切,搜库参数碎片离子质量容许误差:0.02da,母离子质量容许误差:7ppm,允许三种可变修饰:oxidation(m)15.99、acetylation(protein n-term)42.01、deamidation(nq)0.98,且所有肽段经过-10logp≥20质控过滤;
40、3.2.2)de novo peptide分析:
41、使用了三种方法对de novo only peptide进行了去冗余和基因组定位,首先使用orffinder对s187全基因组和s187转录组测序得到的全部转录本的核苷酸序列进行六帧翻译建库,通过肽谱匹配软件peptidemapper,将de novo only的所有肽段映射到两种蛋白质库中,从映射到数据库中的肽段中筛选对应100aa以下的orf的肽段,并将其再提交到uniprot的peptide search工具进行搜索;参数设置如下:搜索物种限定为actinobacteria,并将亮氨酸和异亮氨酸视为等价物,由此过滤掉uniprot数据库中已报导的蛋白中存在的肽段;通过肽段对应的orf id进一步确定剩余新肽段的基因组位置,并统计其所在的orf位置坐标,肽段与对应orf的起始密码子和终止密码子的距离,筛选得到sep,其中具有平均局部置信度≥80%且没有后修饰的肽段作为高可信度的新肽段。
42、于本发明的一种实施方式中,在步骤3.2.2)中采用另一种novel peptide分析,首先通过blastp比对nr数据库中的放线菌序列,task设置为blastp-short,打分矩阵设置为pam30,e-value设置为10,从而删除de novo肽段的冗余部分;后续使用tblastn与s187基因组进行比对,设置pam30评分矩阵并寻找最佳匹配;匹配中筛选80%以上相似度和覆盖率以及e值≤1的肽段,随后根据肽段在基因组定位使用python脚本进行orf批量查找,以起始密码子atg、gtg、ttg、ctg到终止密码子tga、tag、taa,筛选长度小于100个氨基酸的seps;使用在线工具blastp与ncbi nr数据库过滤数据库中注释过的同源序列;最后,选择具有长度≥7aa,和基因组比对错配数不多于2个氨基酸的肽段序列作为novel seps。
43、于本发明的一种实施方式中,步骤3)还包括以下步骤:
44、3.3)ncrna预测:通过将s187转录组测序质控后的clean reads映射回基因组,使用cufflink或stringtie进行依据参考基因组注释文件转录组组装;并使用cuffcompare将组装好的转录本与基因组注释文件进行比对,根据基因组位置信息对新转录本进行分类,对新转录本和ncrna进行了预测。
45、于本发明的一种实施方式中,步骤3)还包括以下步骤:
46、3.4)链霉菌s187与模式菌株共有seps预测:
47、3.4.1)模式菌株转录组数据分析
48、选取sra公共数据库中的链霉菌属模式菌株天蓝色链霉菌的转录组数据,采用以下分析方法进行处理:
49、质控:首先使用软件fastqc和trimmomatic对raw data的reads进行数据质控,通过检测则作为clean data进行后续分析;
50、clean data映射回基因组:使用两种转录组分析流程进行了转录组分析,分别用到了bowtie2和star软件进行reads mapping,首先需要根据天蓝色链霉菌参考基因组fasta文件分别构建两软件的索引文件,再根据索引文件使用bowtie2和star两软件对fastq文件中的clean reads进行映射回贴到参考基因组;将得到的sam文件通过samtools转换和排序,依次生成二进制的bam文件和根据基因组顺序排序后的sorted.bam;使用基因组浏览器igv打开排序后的bam文件,可用来可视化测序reads和基因组以及注释信息等的分布情况;
51、转录本定量和smorfs筛选:对应于bowtie2和star的reads映射回基因组,分别使用了featurecounts工具和rsem软件依据天蓝色链霉菌基因组注释文件分别进行了转录本的定量;得到clean data中对应基因片段的reads的count数,由count数先标准化测序深度,再进行基因长度的标准化,得到fpkm,即每百万reads中该基因的每千碱基长度对应的reads数,来估算基因的表达水平;普遍将fpkm大于1作为该基因稳定转录的必要条件,筛选7组转录组数据中转录本长度小于等于303nt,且fpkm均大于1的作为天蓝色链霉菌中的smorfs;
52、3.4.2)共有seps和s187特有seps预测:
53、使用emboss中的transeq工具将天蓝色链霉菌转录组smorfs翻译成氨基酸序列,生成seps氨基酸序列;将其作为query,使用blastp与基因组smorfs预测所建的s187基因组smorfs全库氨基酸序列进行比对,同时使用tblastn与s187全基因组核苷酸序列进行比对,e-value设置为1e-5;成功参与匹配的seps作为两株菌共有seps进一步提交到cd-search在线工具进行保守结构域查找和功能位点预测,同时具备两特征的seps将作为更可能真实存在和发挥功能的小蛋白参与后续的分析;
54、使用blastp对天蓝色链霉菌和s187两株菌共有的seps和126个db search鉴定到的seps进行比对,参数选择evalue为1e-5,输出格式为outfmt 6,将没有参与匹配的小蛋白作为s187特有的seps。
55、综上所述,本发明提供一种海洋链霉菌s187中小蛋白预测和鉴定方法,本发明的有益效果是:。
56、根据基因组序列信息对smorfs的预测,本发明找到了较为全面的以完整orf(开放阅读框)为基础的基因组smorfs库,以为后续的分析垫定基础。共线性关系也说明s187与模式菌株存在着较为接近的进化关系,因此链霉菌属模式菌株天蓝色链霉菌中较为成熟的研究对s187的研究和smorfs挖掘具有重要的参考意义。
57、进一步地,本发明将六帧翻译理论酶切序列库或打碎生成肽段,而后对肽段理论二级谱图和实际谱图进行匹配,根据dda数据鉴定到肽段的二级谱图和保留时间等信息进行谱库的构建。后续进行dia质谱上机检测和谱图解析,利用dda数据生成的谱库,获取库中每个肽段对应的碎片离子特征,以肽段为中心对dia数据进行靶向信息提取和肽谱匹配打分,并基于二级谱图匹配以谱图为中心进行谱库搜索,从而完成dia二级谱图的多肽鉴定,提高鉴定的灵敏度和精确度,且搜索速度更快。
58、进一步地,对数据库搜索鉴定到的肽段进行了拼接,统计seps中肽段平均覆盖率为39.77%,而且在其中有四个全长蛋白被鉴定到。说明了不使用酶切的自上而下的小蛋白鉴定策略可能有助于在本发明的多肽测序中测到氨基酸长度更长的多肽和提高蛋白对应多肽的覆盖率,为后续分析提供更可靠的肽段定位和定量信息。
59、进一步地,在基因注释水平有限的情况下依赖数据库搜索鉴定肽段仍存在一定的局限性。而de novo测序鉴定到的新肽段,为基因注释信息的补充提供了翻译层次的证据,补充了db search的不足。使用公共数据库模式菌株转录组数据对非模式菌株中的小蛋白识别具有实际的参考意义。
- 还没有人留言评论。精彩留言会获得点赞!