一种基于基因调控网络识别驱动调控因子的方法
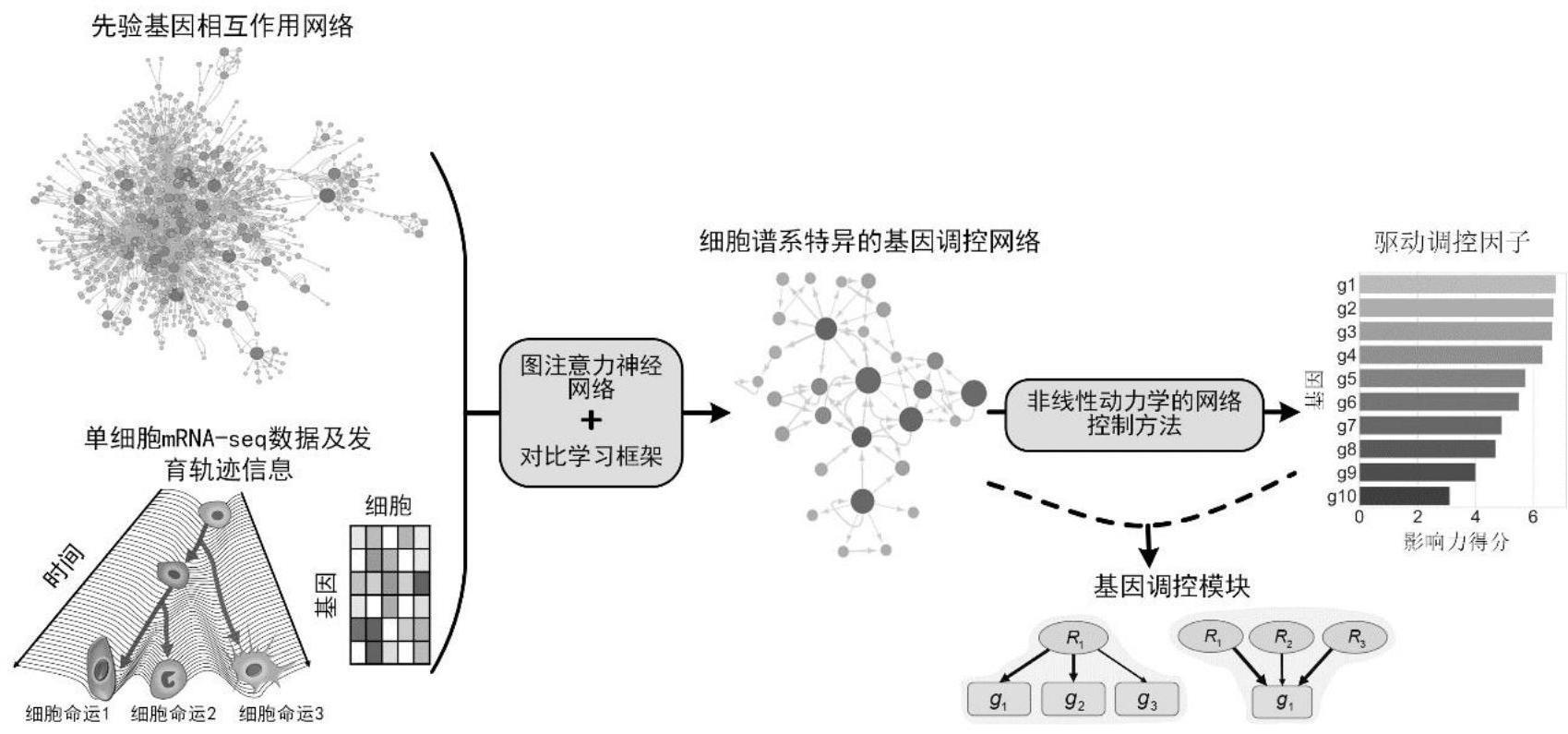
本发明涉及生物信息学领域,具体涉及一种基于基因调控网络识别驱动调控因子的方法。
背景技术:
1、细胞命运决定的机制关系到人体发育、维持稳态、癌症等疾病的发生发展。细胞通过分化产生功能各异的细胞类型,如骨髓造血干细胞受到激素刺激后可分化为淋巴干细胞,并进一步选择性分化为t淋巴细胞和b淋巴细胞。此外,细胞中基因变异等众多复杂因素,可导致正常细胞转变为癌细胞,使细胞命运发生根本性的变化。细胞的分化和重编程等过程是由复杂的基因调节控制的,每个细胞通过整合大量的信号并且执行复杂的基因调控变化来决定自己的命运。究竟什么因素决定了细胞命运,一直是生命科学领域一个基础且非常重要的问题。
2、从基因到细胞层面阐明细胞命运决定机制至关重要,随着单细胞测序技术的快速发展,越来越多的研究已经从细胞“群体”层面具体到单个细胞水平上,使得生命科学领域的研究更加精准化,然而通过计算方法从单细胞测序数据中挖掘基因调控关系,并检测决定细胞命运的关键调控因子,仍然是一个巨大的挑战。
3、大量研究发现,细胞命运选择是由一些关键的转录因子调控的,但是这些转录因子如何决定细胞的分化进程还并不完全清楚。很多计算研究通过重建转录因子驱动的基因调控网络来描述细胞命运的转变,这些最关键的转录因子通常被称为主要调控子(masterregulators,mrs),主要调控子被认为负责控制相关表型下的细胞的整个调控程序。目前常用的主要调控子识别方法包括viper,ananse,scenic等方法,其中只有scenic是专门针对单细胞转录组数据设计的。然而,由于单细胞测序数据存在噪声高、缺失值较多等问题,当前基于单细胞转录组测序数据的基因调控网络的构建还不理想,并且目前缺乏专门针对单细胞数据的控制细胞命运决定的主要调控因子识别方法。一般来说,这些主要调控因子仅仅被局限为转录因子,且很难明确哪些调控因子是真正起到驱动作用的,因而迫切需要基于单细胞转录组测序数据的基因调控网络的构建以及对驱动调控因子准确高效地识别。
技术实现思路
1、本发明旨在至少在一定程度上解决相关技术中的技术问题之一。为此,本发明的一个目的在于提供一种基于基因调控网络识别驱动调控因子以及由驱动调控因子控制的调控基因模块的方法。本发明提供的方法充分利用单细胞转录组数据的特征,构建更加精确的基因调控网络,并且基于此网络获得起关键调控作用的驱动调控因子,有助于解析细胞命运决定的调控机制。
2、为此,本发明第一方面提供一种构建基因调控网络的方法,所述方法包括以下步骤;
3、s01:获取细胞内部基因相互作用网络作为背景基因网络;
4、s02:获取单细胞转录组数据,对所述数据进行预处理,获取单细胞发育轨迹信息;
5、s03:基于图注意力神经网络构建所述基因调控网络的编码器;
6、s04:输入所述背景基因网络和所述单细胞发育轨迹信息,基于图对比学习框架对所述编码器进行训练;
7、s05:获取基因相互关系,利用注意力系数为所述基因相互关系赋权;
8、s06:设定基因相互关系权重阈值,选取高于所述阈值的基因相互关系,得到所述基因调控网络。
9、其中背景基因网络作为先验网络,用来从中选择与特定细胞命运相关的基因相互作用。单细胞转录组数据进行预处理后获取细胞的不同发育轨迹信息和伪时间信息,将先验网络同上述信息输入基于图注意力神经网络构建的编码器中,并基于图对比学习框架对编码器进行训练,构建得到细胞谱系特异的基因调控网络。该基因调控网络还为后续驱动调控因子的识别以及调控基因模块的识别提供了有力的基础。
10、根据本发明的实施方案,步骤s01中所述细胞内部基因相互作用网络来自nichenet(https://github.com/saeyslab/nichenetr)、omnipath(https://omnipathdb.org/)、inbiomap(https://inbio-discover.com/)或pathwaycommons(https://www.pathwaycommons.org/)中的至少一个,其中所述nichenet、omnipath、inbiomap和pathwaycommons剔除掉细胞间的配体-受体相互作用关系,将无向边处理为双向边,由此获得有向的基因关系网络。
11、根据本发明的实施方案,所述步骤s02中所述预处理包括:
12、基于所述单细胞转录组数据中每个细胞表达的基因数量对细胞进行过滤,根据每个基因参与表达的细胞数目对基因进行过滤,去除低质量细胞和基因后,对基因表达量进行归一化处理,获取单细胞发育轨迹信息。
13、根据本发明的实施方案,所述单细胞发育轨迹信息包括采用slingshot、dpt、palantir中的至少一种方法获取。
14、根据本发明的实施方案,步骤s03中所述编码器包括:注意力机制函数、批量归一化、前馈神经网络和激活函数。其中,输入的先验基因网络被表示为一个有向图g=(v,e),其中节点v(|v|=n)表示基因,e表示边。图g的邻接矩阵表示为其中aij=1表示有一条从vi到vj的边,否则aij=0。
15、根据本发明的实施方案,所述编码器的组成方式包括:每一层图注意力神经网络之前进行所述批量归一化,每一层图注意力神经网络之后连接所述前馈神经网络。
16、根据本发明的实施方案,所述组成方式堆叠两层。
17、根据本发明的实施方案,所述注意力机制函数包括如下公式:
18、
19、
20、
21、其中,att函数为注意力函数,度量节点vi和节点vj间的相关性;l表示所述图注意力神经网络中的层;和分别为与源节点和目标节点相关的权重矩阵;和)分别为节点vi和节点vj的基因特征表示,其中对于第一层为输入的基因表达谱数据,dscore为基因差异表达得分;
22、eij通过softmax函数归一化得到注意力系数αij;τ为温度参数,且τ<1;
23、最终节点vi的输出特征是得到的注意力系数αij所对应的特征的线性组合,使用多头注意力机制同时捕捉多个表征子空间的基因特征表示得到σ是gelu激活函数;k是多头注意力的总头数;||表示连接操。
24、基于注意力机制的模型,通过对节点特征之间的相关性进行评分,来学习每个节点的邻居对其特征表示的重要性。节点vi和节点vj间的相关性可以用一个注意力函数att来度量,后通过softmax函数归一化得到一个概率值,即注意力系数αij,最终每个节点的输出特征是上述归一化注意力系数所对应的特征的线性组合,并使用多头注意力机制进行拼接。
25、在注意力函数att中,使用了基于余弦相似性的注意力得分函数,以更直接地考虑节点特征之间的相似性,即只考虑基因调控关系的强度,而忽略调控作用是否为激活或者是抑制的。同时此步骤考虑了基因在细胞命运变化过程中起始和结束状态之间的差异表达,如下式所示:
26、
27、dscore∈[0,1]为基因差异表达得分,用来放大细胞发育过程中显著变化的基因之间的关联性。
28、此外在softmax函数中使用了温度参数τ。当τ>1时,越大的值会导致更均匀的概率;而当τ<1时,越小的值会导致更有区分度的概率。因此此处限定τ<1使得概率分布更加尖锐,有助于注意力集中于少数更相关的邻居节点上。
29、根据本发明的实施方案,所述批量归一化包括采用如下公式对节点vi的基因表达谱进行处理:
30、
31、其中,bn为batch normalization函数。
32、根据本发明的实施方案,所述前馈神经网络包括如下函数:
33、
34、其中,w1和w2为系数矩阵,b1和b2为偏置项。
35、根据本发明的实施方案,所述激活函数如下式所示:
36、
37、根据本发明的实施方案,所述步骤s03进一步包括:将背景基因网络划分为传入网络(incoming network)和传出网络(outgoing network),所述传入网络和传出网络上的计算同时进行,根据以下公式将节点vi的基因特征表示连接起来:
38、
39、其中,和分别表示传入网络和传出网络上的基因特征表示,concat表示拼接操作。
40、背景基因网络的划分可分别考虑在图神经网络中不同方向的消息传递,有助于理解基因作为调控因子或被调控目标的重要性,由此可提高注意力的可解释性,使其更具有生物学意义。
41、根据本发明的实施方案,所述基因差异表达得分dscore的计算方式如下:
42、
43、其中,lx表示基因x的表达量的log2fc,c和d是标量参数,所述c和d分别在每一层图注意力神经网络上具有相同的数值。
44、根据本发明的实施方案,步骤s04中所述图对比学习框架包括采用深度图信息最大化算法,所述深度图信息最大化算法的损失函数如下式所示:
45、
46、其中,n是所述图注意力神经网络中的节点数目,e表示步骤s03中的编码器,(x,a)是一对真实的节点特征和网络结构,是通过随机打乱网络而得到的作为负样本的节点特征和网络结构,si代表全局的图级别的表示,表示被扰动后的嵌入向量,是一个权重矩阵,σ是sigmoid激活函数。
47、深度图信息最大化算法(deep graph infomax,dgi)通过最大化局部图结构和整个图的全局表示之间的互信息来对比地获得网络中节点的表示。所述dgi的损失函数被定义为二元交叉熵损失。
48、根据本发明的实施方案,所述si的计算方式如下:
49、
50、根据本发明的实施方案,步骤s05中为所述基因相互关系赋权包括利用如下函数进行赋权:
51、
52、
53、
54、其中,aij表示邻接矩阵,aij=1表示有一条从节点vi到节点vj的边,否则aij=0;和分别为由传入网络和传出网络上计算得到的注意力系数;。
55、在编码器模型收敛后,使用注意力系数来度量基因之间的相关性的强弱,本发明通过将注意力系数分别与中心节点在传入网络的入度相乘或在传出网络中的出度相乘来对其进行缩放,最终的相互作用权重βij是两个方向网络上缩放注意力系数的平均值。当模型采用两层图注意力神经网络时,最终通过将两层的缩放注意力系数结合起来。
56、根据本发明的实施方案,步骤s06中所述设定基因相互作用关系权重阈值的方法包括:选择阈值使得构建的基因调控网络中边的数目为基因数目的k倍,其中k为用户设定的参数,该参数k即为所构建的基因调控网络的平均度,一般k为选自5~12的任一整数,k越大网络越稠密。
57、本发明第二方面提供一种驱动调控因子的识别方法,所述识别方法包括以下步骤:
58、s07:基于基因调控网络识别驱动基因,得到候选驱动基因;
59、s08:计算所述候选驱动基因的影响力得分,并根据所述影响力得分排序;
60、s09:挑选出影响力得分排名前n的基因作为驱动调控因子;
61、其中,所述基因调控网络通过第一方面所述的方法构建;
62、n为选自1-200的任一整数。
63、基于本发明第一方面提供的基因调控网络构建方法构建基因调控网络,进而识别驱动基因,通过定义一个关于基因在调控网络中重要性的影响力得分,识别细胞命运决定的驱动调控因子。
64、根据本发明的实施方案,所述步骤s07进一步包括:采用最小反馈节点集和最小支配节点集分别识别驱动基因。
65、最小反馈节点集(mfvs)和最小支配节点集(mds)属于网络控制理论方法。其中,反馈节点集是指图中的一个节点集合,使得去除该集合后的图没有反馈回路。支配节点集是指图中的一个节点集合,使得每个支配集以外的其它节点都是支配集成员的邻居。
66、根据本发明的实施方案,步骤s07中所述候选驱动基因为通过最小反馈节点集和最小支配节点集识别到的驱动基因的并集。
67、根据本发明的实施方案,步骤s08中所述影响力得分的计算方式如下:
68、
69、
70、
71、其中,和分别为传入网络和传出网络上的候选驱动基因影响力得分,isi为最终的候选基因影响力得分;和分别表示节点vi的前驱邻居集合和后继邻居集合;和分别为传出网络和传入网络上的缩放注意力系数。
72、根据本发明的实施方案,本发明第三方面提供一种由驱动调控因子控制的调控基因模块的识别方法,所述识别方法包括以下步骤:
73、s10.分别从传入网络和传出网络中识别驱动调控因子及其邻居基因构成的调控基因模块;
74、其中,所述驱动调控因子通过第二方面所述的识别方法识别得到。
75、根据本发明的实施方案,步骤s10中所述调控基因模块包括:i)在传入网络中,所述调控基因模块包括一个驱动调控因子和所述驱动调控因子的靶基因;ii)在传出网络中,所述调控基因模块包括一个驱动调控因子及调控所述驱动调控因子的其它调控基因。
76、根据本发明的实施方案,所述识别方法进一步包括:
77、s11.对所述调控基因模块在每个细胞中的相对活性进行度量,识别不同细胞类型或状态下具有活性的基因模块。
78、根据本发明的实施方案,步骤s11中所述相对活性的度量方法包括采用aucell、gsea或pagoda2中的至少一种方法。
79、aucell方法是scenic方法中提出的基于细胞全基因组排名中曲线下面积(auc)来计算输入基因集的活性评分指标,可以用它来识别活性较高的细胞状态相关的基因模块。
80、gsea方法是一种基因集合分析方法,通过比较一个预定义的基因集合在不同条件下的基因表达谱,来评估该基因集合在某一特定条件下的富集程度。
81、pagoda2方法对每个细胞拟合一个误差模型,对细胞中每个基因的残差进行重新规范化,每个基因组的评分矩阵由其第一个加权主成分进行量化。
82、本发明相对于现有技术具有以下有益效果:
83、本发明提供的方法基于单细胞转录组测序数据构建了基因调控网络,通过该网络可高效准确识别控制细胞命运决定的主要调控因子和调控基因模块。本发明的方法准确可靠,可应用到多领域中,通过预先计算获得的基因调控关系及关键的调控基因等信息可以为实验人员提供参考,节省生物实验成本,提高环保效益;也可应用到疾病治疗中,筛选出的影响疾病发生发展的重要基因及其调控关系,为医生诊断提供帮助,并且能够提高药物治疗靶点筛选的效率等,具有广阔的前景。
84、本发明的附加方面和优点将在下面的描述中部分给出,部分将从下面的描述中变得明显,或通过本发明的实践了解到。
- 还没有人留言评论。精彩留言会获得点赞!