一种关于大型人群队列多性状的多位点关联分析方法
本发明涉及遗传算法,应用于流行病与卫生统计,尤其涉及一种关于大型人群队列多性状的多位点关联分析方法。
背景技术:
1、现有关联的分析方法主要是基于单性状与多位点模型,或者多性状的单位点模型,特别地,仅适用于样本量适中或较小的情形,缺少针对大型人群队列生物银行数据,利用多位点模型进行多性状联合分析的研究。当数据的样本量和标记数目都非常巨大,在进行多重和多元线性回归分析时,面临的主要问题有:①基因型矩阵太大,直接计算会超出内存大小;②将一元分析方法简单的拓展到多元分析,计算复杂度会大大增高,一些近似策略在多元分析时不成立;③很多程序由于未考虑数据在内存和硬盘交换的时间消耗,分析大数据不可行;④大部分软件仅利用cpu多线程加速,未考虑gpu并行设计,计算效率有很大提升空间;⑤很多软件局限于某一特定操作系统,运行环境的搭建十分复杂。
2、对于上述相关的技术问题,申请人在前期工作中提出了基于线性混合模型的多位点gwas两阶段方法scoreeb和hrepml,与广泛使用的单位点方法gemma进行了比较,模拟数据显示多位点方法的统计功效都要优于gemma。虽然scoreeb和hrepml相比其它多位点gwas方法在运算速度方面得到提升,遗憾的是,它们仍然局限于中小样本,在分析诸如ukbiobank等大型人群队列数据时计算效率太低。目前,针对百万大型人群队列,尚未有学者提出高效的多性状多位点关联分析方法。
技术实现思路
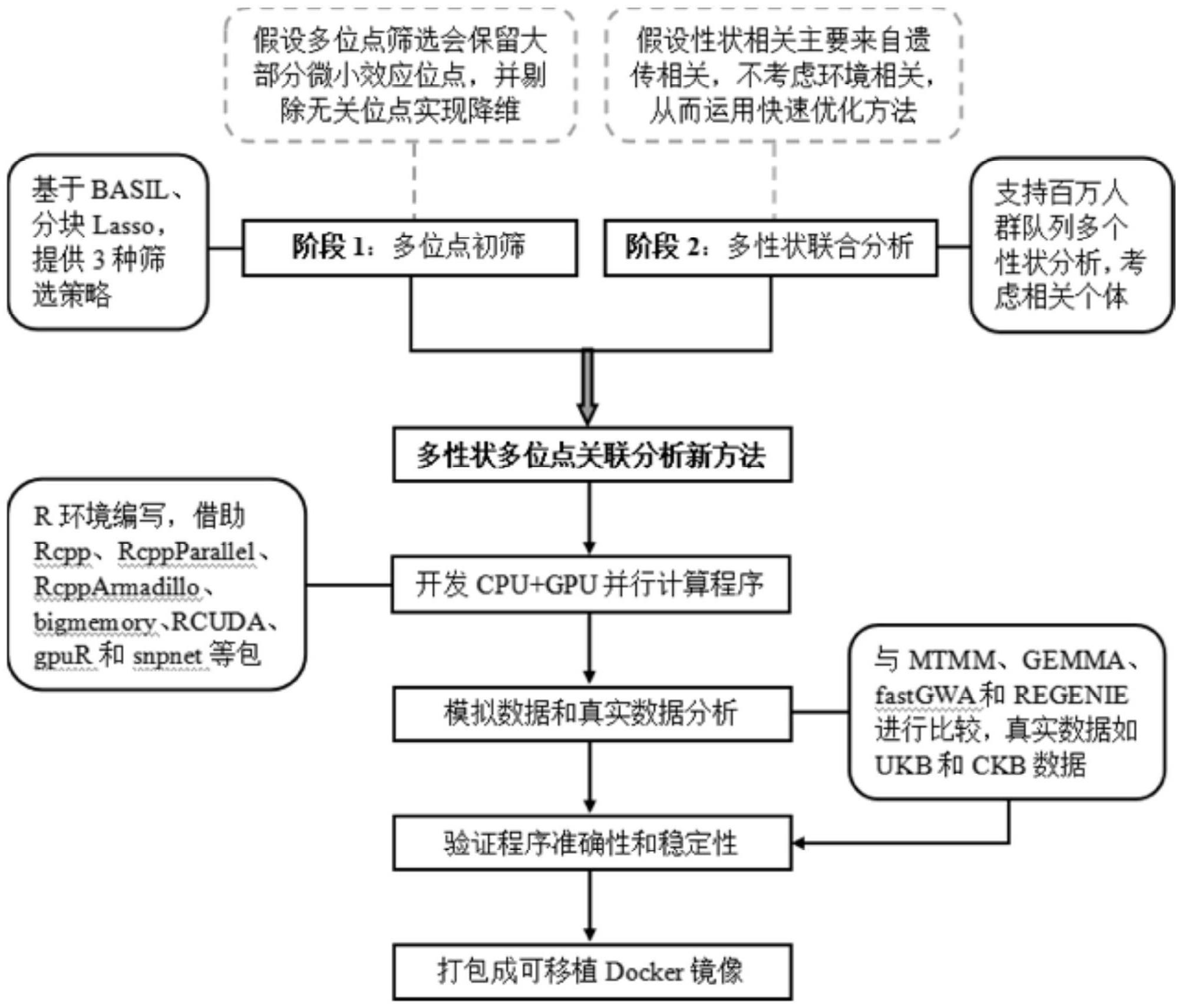
1、本发明的目的是为了解决现有技术中缺乏高效的多性状多位点关联分析方法的技术问题,本技术中建立了适用于百万人群队列的多性状多位点全基因组关联分析新方法,研制新方法的docker可移植并行计算软件包,其目的是构建适用于百万人群队列的多性状多位点全基因组关联分析技术平台,为探索复杂性状或疾病的微小效应和一因多效变异位点(或基因)提供新技术。
2、为了实现上述目的,本发明采用了如下技术方案:
3、一种关于大型人群队列多性状的多位点关联分析方法,采用“多位点初步筛选→多性状再次筛选”两阶段分析的策略,第一阶段建立多位点线性回归模型,探索求解系数的压缩估计方法;第二阶段建立多性状线性混合模型,提出性状间的相关主要来源于遗传相关的假设,基于此假设通过矩估计、hutchinson估计和预条件共轭梯度下降法等数值优化技术快速参数估计值来获得检验统计量。
4、优选的,多位点初步筛选包含以下步骤:建立多重线性模型:
5、y=cα+xβ+ε
6、上述公式中,为性状列向量,n为样本含量;为校正矩阵,为其相应的效应向量;为基因型矩阵,q为标记的个数,为标记效应列向量,为随机误差项;
7、对多重线性模型进行变量选择,即求解下列等式的最小值:
8、
9、上述公式中,||·||w表示w范数,λ为调节参数;
10、对d个复杂性状(d≥2)进行多位点初步筛选,并对筛选结果取并集,x∪=xs1∪…∪xsd表示多位点初筛得到的基因型矩阵,其中,xs1(1≤i≤d)表示第i个性状得到的基因型矩阵。
11、优选的,所述s1中多位点初步筛选的方法为下列方法之一:
12、①全局批量筛选迭代lasso的方法basil,通过r语言软件包snpnet实现;
13、②通过plink 2.0进行快速的单位点筛选,将p<10-3的位点保留(该阈值可根据实际数据灵活设定),用xs表示保留的基因型子集矩阵,通过r语言软件包biglasso对xs子集进行多变量筛选;
14、③将巨大的基因型矩阵分为m块,保证每块子矩阵小于计算机内存,用biglasso分别对m个子矩阵进行多变量选择,汇总所有结果构成基因型矩阵x′s,对x′s再运行一次lasso。
15、优选的,多性状再次筛选包含以下步骤:建立多元混合线性模型
16、
17、上述公式中,为多性状矩阵,d为性状个数;为校正矩阵,为其固定效应,x∈x∪为n维当前检验标记基因型列向量,为标记效应行向量,β~n(0,vβ),为克罗内克积;和分别表示随机多基因效应和剩余误差矩阵,和服从多元正态分布,k为遗传关系矩阵,in为单位阵,和为遗传协方差和剩余协方差;
18、y服从多元正态分布:
19、
20、令θ=(α,vβ,vg,ve),则关于y的对数似然函数可以表示为:
21、
22、上述公式中,,vec(·)表示将矩阵的所有列堆在一起后形成的列向量;
23、然后对参数θ进行估计,进一步获得零假设下的统计量和p值,h0:β=0,即所有性状的标记效应大小为0,0表示d维零向量,备择假设h1:β≠0;因为β~n(0,vβ),所以,对β的假设检验亦可通过对vβ进行,即h0:vβ=0,h1:vβ≠0;
24、设性状之间的相关主要有遗传相关决定,不考虑环境相关,此时,
25、
26、的估计可由第1到d个性状分别建立线性方程以并行的方式获得,yi=cai+xβi+ξi+εi(1≤i≤d),各参数与前面所述含义相同,在零模型下的矩估计:
27、
28、构造hutchinson估计量可以快速获得近似解:
29、
30、上述公式中,z1,…,zb是b个独立的随机列向量,服从均数为0,协方差为in的正态分布;
31、性状i,j(1≤i,j≤d,i≠j)间的遗传相关系数可通过下式得到:
32、
33、由式子(3)到(6),可以获得vg,ve的估计值以及零模型下v的估计值
34、α的估计值构造检验统计量:
35、
36、统计量s中,因此,统计量s在零假设下可表示为:
37、
38、将的估计值代入式子中并进行合并化简可得:
39、
40、因此,统计量s可化简为:
41、
42、
43、在零假设下且样本量较大的时候,统计量s渐近服从自由度为d的卡方分布。将和看作一个整体来计算,通过预条件共轭梯度下降法获得其近似值;
44、从x∪中随机选择l个标记(如l=50),此时,对l个标记分别进行如下计算:
45、
46、mean表示对向量各元素相加并求其算术均数,则统计量s可近似为:
47、
48、本技术拟采用“多位点初步筛选→多性状再次筛选”两阶段分析的策略:首先,采用大型数据lasso求解器对原始数据进行降维;然后,建立多元线性混合模型,巧妙的利用矩估计、hutchinson估计和预条件共轭梯度下降法等数值优化技术快速参数估计来获得检验统计量。本技术的上述方法能够发现更多微小效应和一因多效位点,对解析复杂性状或疾病遗传机理具有重要意义,并将其应用于uk biobank、china kadoorie biobank等大型队列gwas数据,以期解密基因和人类疾病的关系,为人类疾病的预防和治疗提供帮助。
- 还没有人留言评论。精彩留言会获得点赞!