基于基因组突变特征及基因集表达特征的卵巢癌HRD分型系统及方法与流程
本发明涉及基因检测,具体地说,是关于基于基因组突变特征及基因集表达特征的卵巢癌hrd分型系统及方法。
背景技术:
1、卵巢癌(ovarian cancer)是恶性程度极高的妇科肿瘤[1]。70%患者一经发现即为晚期。新诊断的晚期卵巢癌(iiib-iv期)的标准治疗包括初次减瘤手术后进行紫杉醇和卡铂辅助化疗,或新辅助化疗后间歇性减瘤手术后进行辅助化疗[2-4]。尽管大多数患者通过任一方法获得完全缓解,但仍有60-75%的患者在2-3年内复发。晚期卵巢癌总体5年生存率仍然不足30%[5]。
2、同源重组修复(homologous recombination,hr)是dna双链损伤修复的重要途径,如果异常会导致dna双链损伤修复缺陷,即hrd,大约有53%的卵巢癌患者存在同源重组修复缺陷(homologous recombination deficiency,hrd)。多腺苷二磷酸核糖聚合酶(polyadp ribose polymerase,parp)抑制剂维持治疗是近年卵巢癌治疗史上的一次革命[6],尤其针对brca基因致病突变和hrd的卵巢癌患者,通过抑制肿瘤细胞dna的修复,通过合成致死作用促进肿瘤细胞凋亡,能够延长这类患者的复发时间,从而改善预后。hrd作为parp抑制剂敏感的生物标志物已用于临床伴随诊断,成为临床精准用药的分子靶点。
3、目前,hrd评估主要有2个技术路线。第一个方法是检测hr通路中相关基因的突变。hr是一条涉及到多个步骤的复杂的信号转导通路,其中关键蛋白为brca1和brca2,携带brca1和(或)brca2基因的胚系有害突变的人群其一生中患乳腺癌、卵巢癌、前列腺癌、黑色素瘤和胰腺癌的风险增加[7],随着研究的深入,不断有新基因被发现参与hr作用,成dna损伤修复的复杂系统,这些蛋白包括atm、rad51、palb2、mre11、rad50、nbn和fa蛋白等[8]。利用二代测序技术,可以评估许多hrr基因是否存在胚系或体系突变,但对于基因突变的注释仍然是一大挑战;此外,由于表观遗传修饰等因素,单纯检测hr基因的突变不能完全反映患者的hrd状态。
4、第二个方法是通过检测hrd产生的结果,即检测基因组瘢痕(genomic scars)。hrd的功能学评估有助于弥补检测hr基因突变的不足,当hrd存在时,基因组变异累积,等位基因失衡可能导致“基因组瘢痕”,通过对“基因组瘢痕”所表示的hrd进行评估可以不考虑潜在的遗传或表观遗传机制[9]。可使用hrd相关基因的测序芯片或全基因组范围内的snp算法。基因组瘢痕检测目前仅有2个fda批准且经过临床验证的产品,即foundationfocustmcdxbrca loh和myriadcdx。前者通过检测brca突变状态和基因组杂合性缺失(loss ofheterozygosity,loh)而进行综合评估,后者通过检测基因组不稳定性状态的3项指标(loh、端粒等位基因不平衡(telomeric allelic imbalance,tai)、大片段迁移(large-scale transition,lst))从而进行评估。目前国内尚无hrd检测产品获得cfda或伴随诊断批准。
5、在卵巢癌中若仅检测hrr基因(含brca1/2),受益人群比例约为25%,但若进行基因组hrd评分(含brca1/2检测),受益人群比例可提高至50%。目前,基因组hrd评分通常需要大量不相连的snp位点组成panel,snp位点数量常在3万至5万个点。例如,中国专利申请公布cn112226495a公开了一种dna同源重组异常的检测方法,包括:(1)snp位点筛选;(2)为筛选到的snp位点设计捕获探针;(3)基因组dna提取和文库构建;(4)文库靶向富集;(5)高通量测序并分析测序数据,判断hrd状态时使用kolmogorov smirnov检验或者scarhrd。
6、目前已有的hrd产品,主要使用hrd相关基因的测序芯片或全基因组范围内的snp检测,基于靶向测序(target region sequencing),即利用探针杂交的方法对特定位点或区域进行捕获和富集,并进行高通量测序。但是,该方法中芯片或目标捕获区域设计目标范围太窄,只能检测固定位点,存在很大的局限性。同时这类方法的探针设计及确定后的性能验证,还是临床样本的检测,均存在成本高的问题,不能随其他产品而伴随应用。这类方法还只能对肿瘤组织dna及配对的白细胞的dna同时进行检测来进行hrd状态的评估,接受检测的样本类型单一。
7、综上,传统的检测同源重组修复缺陷的方法存在的不足之处在于:只能检测固定位点,不全面,存在较大局限性,并且检测成本高,需要设计特定的探针,且接受检测的样本类型单一。所以亟需一种能低成本、检测较为全面且准确度高的方法用于检测hrd状态。而目前关于如本发明的基于基因组突变特征及基因集表达特征的卵巢癌hrd分型系统及方法还未见报道。
技术实现思路
1、本发明的第一个目的是,针对现有技术中的不足,提供了基于基因组突变特征及基因集表达特征的卵巢癌hrd分型方法。
2、本发明的第二个目的是,提供了一种基于肿瘤组织基因组突变特征及基因集表达特征的卵巢癌hrd分型系统。
3、本发明的第三个目的是,提供了一种卵巢癌hrd分型方法的应用。
4、为实现上述第一个目的,本发明采取的技术方案是:
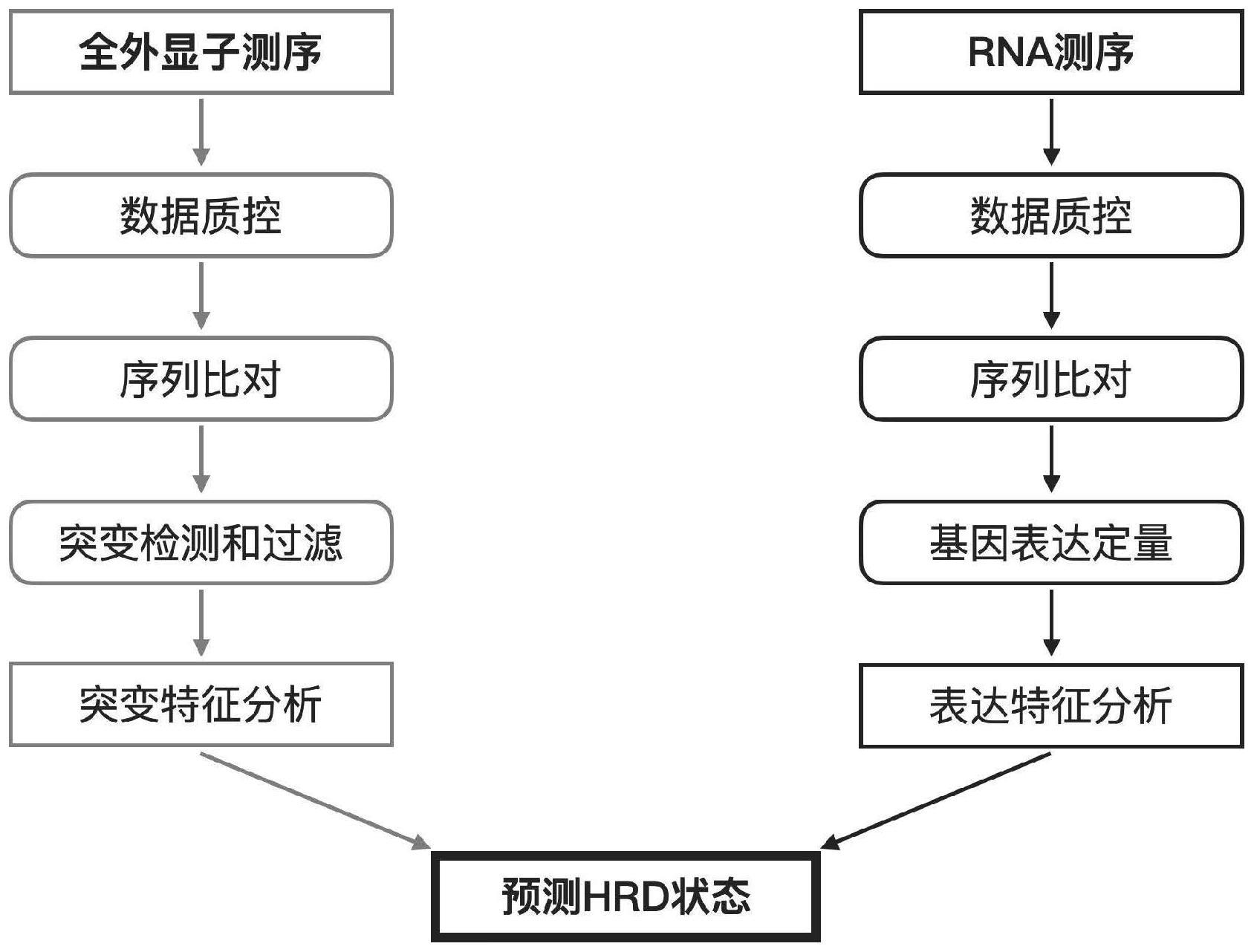
5、一种基于基因组突变特征及基因集表达特征的卵巢癌hrd分型方法,所述分型方法包括基于全外显子测序(wes)数据构建的wes模型,基于转录组测序(rna-seq)数据构建的rna模型,以及结合全外显子和转录组测序数据构建的wes+rna综合模型。
6、作为一个优选例,上述wes模型的建立包括以下步骤:
7、a1:二代测序;
8、a2:数据质控;
9、a3:序列比对;
10、a4:突变检测、过滤和注释;
11、a5:突变特征分析;
12、a6:hrd状态评估。
13、作为一个优选例,上述步骤a1将肿瘤组织的dna样本使用全外显子捕获后进行二代测序;所述步骤a2对原始测序数据(raw reads)中的测序接头、低质量碱基、模糊碱基和长度等进行过滤,得到高质量数据(clean reads)。其中,质控后的长度至少为75bp;所述步骤a3将clean reads与人类全外显子的参考序列进行比对,得到比对文件并去除比对文件中的重复reads;所述步骤a4中的突变过滤包括基于正常样本库(panel of normal)和gnomad数据库(the genome aggregation database)的胚系变异过滤以及基于测序深度、正负链比、变异支持序列数、突变频率、基因组黑名单区域(blacklist region)等的低质量变异过滤。其中,panel of normal基于38个血液样本,并将随着数据的收集定期更新。其中,测序深度需要≥30x。其中,正负链比介于10%-90%。对热点突变snv、非热点点突变snv、热点indel和非热点indel的支持序列数分别需要≥3、8、2、5。其中,热点突变snv、非热点点突变snv、热点indel和非热点indel的突变频率分别需要≥0.01、0.05、0.01、0.03;所述步骤a5中突变特征分析包括突变频谱计算和突变特征指数计算。其中,突变频谱包括单碱基替换(single base substitutions,sbs)突变频谱和小片段的插入和缺失(smallinsertions and deletions,id)突变频谱。其中,突变特征指数基于cosmic数据库收录的各突变特征(signature)对样本突变频谱的贡献率;其中,选定的用于后续hrd状态预测的突变特征包括与hrd及brca突变相关的单碱基替换突变特征3(sbs3)和小片段的插入和缺失突变特征6(id6),以及与非同源性末端接合(non-homologous end joining,nhej)相关的突变特征id8;所述步骤a6中的hrd评估模型包括基于sbs3突变特征指数的判断和基于以id6和id8为参数的逻辑回归模型两个部分。
14、更优选地,基于id6和id8突变特征指数的模型为:
15、η=-0.4261594+4.9839447×id6特征指数+5.8571149×id8特征指数
16、其中,
17、
18、其中,符合下列条件之一的样本将被判定为hrd阳性:(1)sbs3突变特征指数>0.203;(2)wes score>0.832。
19、更优选地,上述rna模型的建立包括以下步骤:
20、b1:二代测序;
21、b2:数据质控;
22、b3:序列比对;
23、b4:基因表达定量;
24、b5:特定基因集表达特征分析;
25、b6:hrd状态评估。
26、更优选地,上述步骤b1将肿瘤组织的rna样本使用polya捕获后进行二代测序;所述步骤b2对原始测序数据(raw reads)中的测序接头、低质量碱基、模糊碱基和长度等进行过滤,得到高质量数据(cleanreads)。其中,质控后的长度至少为75bp;所述步骤b4对原始表达数据(read counts)进行定量归一化,计算基因或者转录本的tpm(transcripts permillion)值;所述步骤b5的目的为筛选与hrd状态相关的基因集;所述步骤b6中的特定基因集表达特征分析包括基于样本在特定基因集的富集程度的表达特征指数计算;所述步骤b7中的hrd评估模型包括基于以特定基因集的表达特征指数为参数的逻辑回归模型。
27、更优选地,上述步骤b5中的基因表达特征分析为对特定基因集的表达特征指数计算;上述特征基因集包括上调基因集与下调基因集,
28、其中上调基因集包含以下基因:
29、ensg00000031544(nr2e3),ensg00000060709(rimbp2),
30、ensg00000074211(ppp2r2c),ensg00000112936(c7),
31、ensg00000118729(casq2),ensg00000124491(f13a1),
32、ensg00000124493(grm4),ensg00000126583(prkcg),
33、ensg00000134569(lrp4),ensg00000135472(faim2),
34、ensg00000135960(edar),ensg00000140297(gcnt3),
35、ensg00000142623(padi1),ensg00000143001(tmem61),
36、ensg00000143171(rxrg),ensg00000143631(flg),
37、ensg00000154263(abca10),ensg00000162344(fgf19),
38、ensg00000163283(alpp),ensg00000165376(cldn2),
39、ensg00000167210(loxhd1),ensg00000167580(aqp2),
40、ensg00000170579(dlgap1),ensg00000171551(ecel1),
41、ensg00000172927(myeov),ensg00000173714(wfikkn2),
42、ensg00000181449(sox2),ensg00000186895(fgf3),
43、ensg00000187537(potem),ensg00000187627(rgpd1),
44、ensg00000196226(hist1h2bb),ensg00000197915(hrnr),
45、ensg00000205038(pkhd1l1),ensg00000205238(spdye2),
46、ensg00000205277(muc12),ensg00000242384(tbc1d3h);
47、其中下调基因集包括下列基因:
48、ensg00000066248(ngef),ensg00000102683(sgcg),
49、ensg00000107317(ptgds),ensg00000110328(galnt18),
50、ensg00000112319(eya4),ensg00000115507(otx1),
51、ensg00000122012(sv2c),ensg00000130700(gata5),
52、ensg00000133937(gsc),ensg00000137878(gcom1),
53、ensg00000147573(trim55),ensg00000158008(extl1),
54、ensg00000159871(lypd5),ensg00000164120(hpgd),
55、ensg00000166106(adamts15),ensg00000166923(grem1),
56、ensg00000168546(gfra2),ensg00000169218(rspo1),
57、ensg00000197467(col13a1),ensg00000198729(ppp1r14c),
58、ensg00000241644(inmt);
59、所述步骤b6中的hrd评估基于以特定基因集的表达特征指数为参数的逻辑回归模型:
60、
61、其中,
62、η=11.90860+14.74870×上调基因集表达特征指数-20.03622×下调基因集表达特征指数
63、其中,符合条件rna score>0.877的样本将被判定为hrd阳性。
64、更优选地,所述wes+rna综合模型包括基于sbs3突变特征指数的判断和基于以wes和rna模型分别计算得到的wes score和rna score为参数的逻辑回归模型两个部分;分析流程包括以下步骤:
65、c1:权利要求2所述的步骤a1-a6;
66、c2:权利要求4所述的步骤b1-b6;
67、c3:hrd评估。
68、更优选地,上述步骤c3中的hrd评估包括基于sbs3突变特征指数的判断和基于以步骤c1和c2计算得到的wes score和rna score为参数的逻辑回归模型两个部分,其中,基于wes score和rna score的模型为:
69、其中,
70、η=-10.470683+9.637483×wes score+7.214411×rna score
71、其中,符合下列条件之一的样本将被判定为hrd阳性:(1)sbs3突变特征指数>0.203;(2)hrd score>0.766。
72、更优选地,上述hrd分型方法基于输入数据类型,预测hrd状态,该模型的准确性估计均使用交叉验证的方法。
73、为实现上述第二个目的,本发明采取的技术方案是:一种基于基因组突变特征及基因集表达特征的卵巢癌hrd分型的系统,其特征在于,所述系统基于上述任一项所述的卵巢癌hrd分型方法;所述系统仅需输入肿瘤样本wes原始测序数据和/或rna-seq原始测序数据;所述系统输出为该样本预测的hrd状态。
74、为实现本发明的第三个目的,本发明采用的技术方案是:上述任一所述的卵巢癌hrd分型系统在制备评估hrd状态的产品中的应用。
75、本发明优点在于:
76、1、本发明提供的一种基于wes测序和/或rna测序评估hrd状态的方法,解决了单一组学数据的局限性和准确度、单一突变类型的局限性以及基于基因差异表达数据易受到批次效应影响的局限性和准确度性,从而扩大受益人群且降低成本,更适合于临床应用。
77、2、本发明基于中国人群构建对照样本库,克服了对血液对照样本的依赖;克服了使用单一组学数据评估hrd状态的局限性,扩大了应用场景;克服了之前判定hrd状态方法中对brca1/2基因突变临床评级注释的依赖;针对中国人群优化hrd分型系统参数;为判断卵巢癌患者的hrd状态,提供了更多的可选方法。
- 还没有人留言评论。精彩留言会获得点赞!