一种基于变分神经网络的疾病预测方法及系统
本发明涉及一种基于变分神经网络的疾病预测方法及系统,属于疾病预测。
背景技术:
1、人类肠道微生物群是一个复杂的微生物生态系统,其中的基因组约为300万,比人类宿主的基因组大150倍。事实上,肠道微生物群通过合成人类基因组无法编码的酶在人体代谢中发挥着重要作用,这些酶可以促进多糖和多酚的分解,促进营养吸收,并提供对病原体的保护。越来越多的研究表明,肠道微生物群的生态失调可能与各种疾病密切相关,特别是那些影响胃肠系统的疾病。为了揭示微生物与人类健康之间的关系,出现了各种组学技术,如宏基因组学、宏转录组学和代谢组学等。每一个都在特定的组学水平上提供了分子机制或生物过程的一种信息。在过去的几年里,越来越多的研究表明,组学数据的组合通常提供了更完整的信息和对微生态学的更好理解,这可以增加人类疾病预测的准确性,提高分析的稳健性,还可以发现重要的生物标志物。值得注意的是,微生物组多组学数据包括各种类型的不同数据,并以其异质性、稀疏性和高维属性而闻名。鉴于这些特点,数据处理需要专门的分析方法,以便于更深入的理解和知识发现。目前高性能机器学习方法在生物领域中受到了相当大的关注,已经开发了大量模型来充分利用多组学信息的潜力。
2、不完整的组学数据在公开的数据库中很常见,这可归因于各种因素,如有限的资金、伦理考虑和隐私问题,这些因素会影响样本的可用性。这给集成分析带来了巨大的挑战。在这种情况下,可以考虑样本丢弃或均值插补。然而,前者将大大减少可用样本的数量,而后者可能会严重扭曲数据的真实分布。现有的利用不完整的多组学数据进行疾病预测的机器学习算法主要存在两个问题,一是无法有效地从高维组学数据中提取相关特征并过滤掉不相关的特征,二是难以在实现不完整多组学数据的灵活集成的同时充分利用其中信息实现高效预测。
技术实现思路
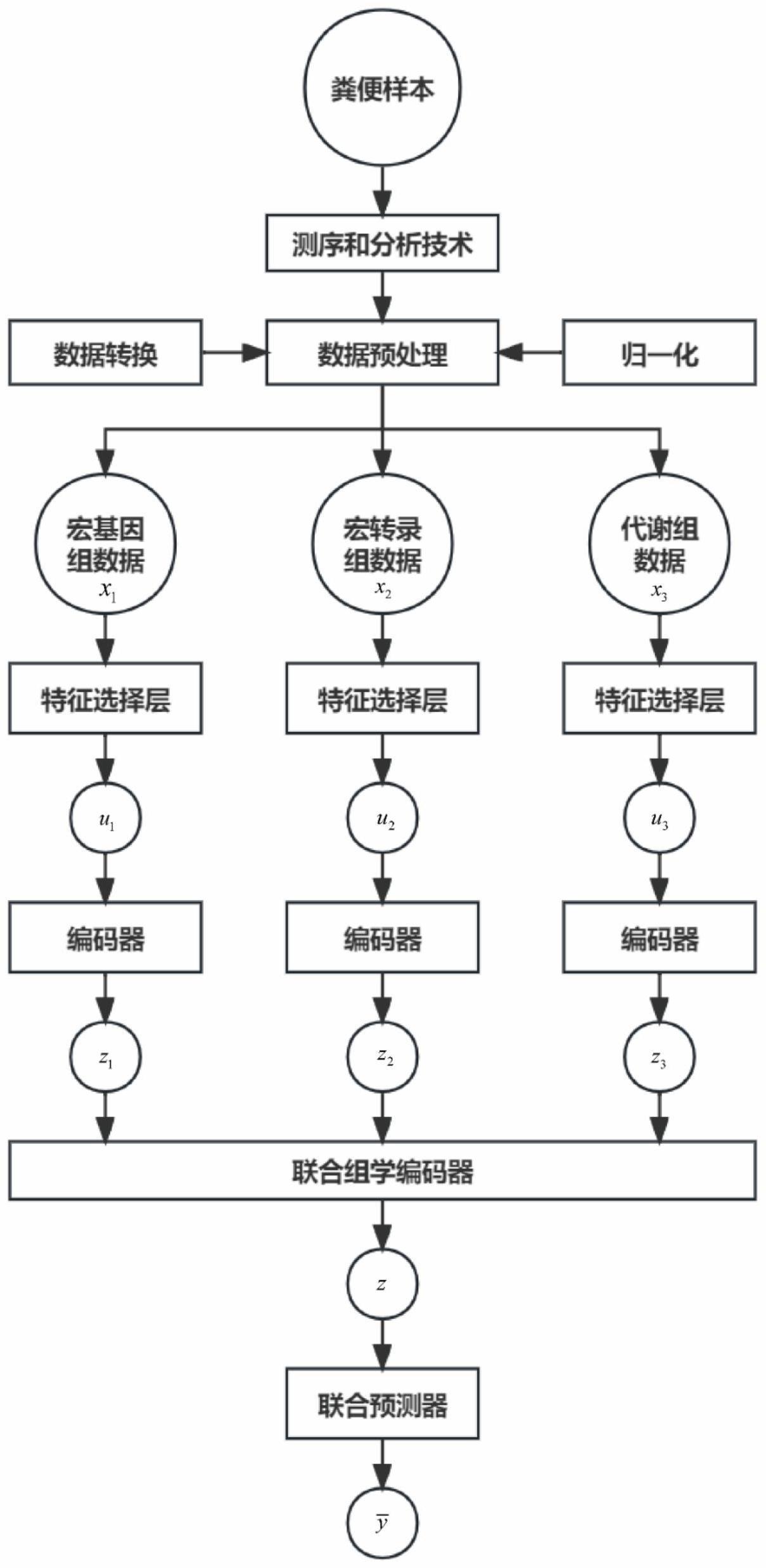
1、为解决上述问题,本发明提供了一种基于变分神经网络的疾病预测方法及系统,该方法基于变分神经网络,利用不完全肠道多组学数据进行疾病预测,收集人体肠道粪便样本,通过测序和分析技术得到样本的菌群丰度数据、通路数据和代谢物丰度数据,对多组学数据进行预处理,将样本拥有的多组学数据代入到训练好的算法框架中,得出生病的概率值,算法框架的输出结果为两类,即患病、未患病。
2、一方面,本发明提供了一种基于变分神经网络的疾病预测方法,包括如下步骤:
3、步骤1:提取样本的dna、rna和代谢物,将dna和rna信息扩增成适合高通量测序的文库,利用高通量测序技术得到测序原始数据,对原始数据进行处理后分别进行物种注释和功能注释得到宏基因组的菌群丰度数据和宏转录组的通路数据,通过质谱分析法得到代谢组的代谢物丰度数据;
4、步骤2:对多组学数据进行预处理,包括数据转换和归一化处理;
5、步骤3:将处理过后的菌群丰度数据、通路数据和代谢物丰度数据代入到训练好的算法框架中,得出生病的概率值,算法框架的输出结果为患病或未患病。
6、在本发明的一种实施方式中,所述步骤2中的数据转换和归一化处理包括如下步骤:
7、步骤2.1:对数据进行以下转换,以使用神经网络进行合理的分析;
8、x=log2(2x+0.00001)
9、其中,x表示菌群丰度数据;
10、步骤2.2:若菌群丰度数据已被转换,则对每一种组学数据均进行以下归一化处理:
11、
12、其中xmean是该组学数据的平均值,xmax是该组学数据中的最大值,xmin是该组学数据中的最小值。
13、在本发明的一种实施方式中,所述步骤3中算法框架利用多组学数据得出生病概率值包括如下步骤:
14、步骤3.1:处理过后的菌群丰度数据、通路数据和代谢物丰度数据编制成三个矩阵,假设代表第v个组学数据的矩阵由n个样本的dv个特征组成,首先每个矩阵通过训练好的特征选择层进行特征选择,计算过程如下:
15、uv=xv·sv
16、其中是训练后逼近one-hot形式的线性变换矩阵,对每个组学数据分别进行特征选择得到uv∈rn×f;
17、步骤3.2:特征选择后的组学数据通过训练好的由全连接层和激活函数组成的编码器,得到每个组学的潜在表示,计算过程如下:
18、
19、μv+∈·σv=zv
20、其中表示多层神经网络的非线性变换过程,∈训练网络时从标准正态分布中随机抽样,模型训练完成后∈固定为0;先由uv得到潜在表示的均值μv和方差σv,再利用重参数化技巧得到每个组学数据的潜在表示zv;
21、步骤3.3:通过使用联合组学编码器简单地整合具有任意缺失情况的不完整多组学数据,并获得联合潜在表示z,计算过程如下:
22、
23、
24、μ+∈·σ=z,∈~n(0,1)
25、其中v代表样本拥有的组学数量,μ0和σ0表示先验分布的均值和方差,∈训练网络时从标准正态分布中随机抽样,模型训练完成后∈固定为0;由已有的组学数据的均值μv和方差σv集成得到联合组学均值μ和方差σ,利用重参数化技巧得到联合组学数据的潜在表示z;
26、步骤3.4:联合组学数据的潜在表示z经过训练好的由全连接层和激活函数组成的联合预测器得出生病概率值计算过程如下:
27、
28、其中fψ表示多层神经网络的非线性变换过程,基于联合组学数据的潜在表示z得到样本是否生病的预测标签。
29、在本发明的一种实施方式中,所述算法框架的训练过程包括如下步骤:
30、步骤s1:收集健康人群和确诊模型预测目标疾病人群肠道粪便样本,对人群进行人工标记,标记生病人的粪便样本为1和不生病人的粪便样本为0,通过测序和分析技术获取样本对应多组学数据,或者搜集公开数据构建多组学数据库,获取有标签粪便样本的多组学数据;
31、步骤s2:对多组学数据进行数据转化和归一化处理;
32、步骤s3:将标注的数据集分为训练集与测试集,利用训练集数据对算法框架进行监督训练,并在测试集上进行测试。
33、在本发明的一种实施方式中,所述步骤s3中算法框架利用训练集数据进行监督训练的一次训练过程包括如下步骤:
34、步骤s3.1:训练集的菌群丰度数据、通路数据和代谢物丰度数据编制成三个矩阵,假设代表第v个组学数据的矩阵由n个样本的dv个特征组成,首先每个矩阵通过特征选择层进行线性变换:
35、te=t0·(te/t0)e/e
36、
37、uv=xv·εv
38、其中,e表示训练总迭代次数,e表示是第几次迭代,t0和te是算法模型的超参数,分别设置为10和0.1,γv为全连接层的参数,ε是从均匀分布(0,1)中随机采样,softmax()表示一种激活函数,经过以上变化由xv得到uv∈rn×f;
39、步骤s3.2:通过特征选择层进行变化的组学数据通过由全连接层和激活函数组成的编码器,得到每个组学的潜在表示,计算过程如下:
40、
41、μv+∈·σv=zv
42、其中表示多层神经网络的非线性变换过程,θv为神经网络的参数,∈从标准正态分布中随机抽样;先由uv得到潜在表示的均值μv和方差σv,再利用重参数化技巧得到每个组学数据的潜在表示zv;
43、步骤s3.3:通过使用联合组学编码器简单地整合具有任意缺失情况的不完整多组学数据,并获得联合潜在表示z,计算过程如下:
44、
45、
46、μ+∈·σ=z,∈~n(0,1)
47、其中v代表样本拥有的组学数量,μ0和σ0表示先验分布的均值和方差,∈从标准正态分布中随机抽样;由已有的组学数据的均值μv和方差σv集成得到联合组学均值μ和方差σ,利用重参数化技巧得到联合组学数据的潜在表示z;
48、步骤s3.4:每个组学数据的潜在表示zv和联合潜在表示z分别经过由全连接层和激活函数组成的预测器和联合预测器得出特定组学预测概率值yv和最终预测概率值计算过程如下:
49、
50、
51、其中和fψ表示多层神经网络的非线性变换过程,和ψ均为神经网络的参数;
52、步骤s3.5:根据模型的损失函数计算损失,进行梯度回传更新模型神经网络的参数,计算过程如下:
53、
54、
55、lt=lj+α∑v∈vlv
56、
57、其中,α和β是结合不同损失的平衡系数,n(μ0,σ0)表示先验分布,n(μv,σv)表示第v个组学潜在表示的分布,n(μ,σ)表示联合潜在表示的分布,kl()表示计算两个分布之间的kl散度即相对熵,y为one-hot形式的真实标签,yv为特定组学预测概率值,为最终预测概率值,n为样本数量,λ为算法框架训练时的学习率,为算法框架中神经网络的参数;如此完成一次模型的训练对神经网络参数进行更新。
58、另一方面,本发明还提供了一种基于变分神经网络的疾病预测系统,应用了所述的一种基于变分神经网络的疾病预测方法,所述系统包括:
59、特征选择层模块,用于选取重要特征对组学数据线性变换;
60、编码器模块,用于将组学数据随机编码成潜在表示;
61、联合编码器模块,用于集成各组学潜在表示;以及
62、预测器和联合预测器模块,用于由编码器模块编码的潜在表示提供标签推断,即疾病预测结果。
63、在本发明的一种实施方式中,所述特征选择层模块为布尔矩阵。
64、在本发明的一种实施方式中,所述编码器模块由全连接神经网络构成。
65、在本发明的一种实施方式中,所述联合编码器模块为计算模块。
66、在本发明的一种实施方式中,所述预测器和联合预测器模块由全连接神经网络构成。
67、本发明提供的一种基于变分神经网络的疾病预测方法及系统,具有以下优点:其一,本发明提出了一个基于变分神经网络的新框架,能够用于整合不完整的微生物组多组学数据,预测疾病和寻找疾病相关的生物标志物。其二,本发明引入了具体分布,可以在每个微生物组学中选择与目标疾病最相关的特征,从而提高模型的可解释性。其三,算法利用信息瓶颈原理构建模型训练的损失函数,促进学习单组学潜在表示和联合组学潜在表示,使得算法具有较高的预测准确率和鲁棒性。其四,本发明对多组学数据的完整性要求不高,可以灵活地利用样本拥有的组学数据进行疾病预测。
- 还没有人留言评论。精彩留言会获得点赞!