判断融合基因真实性的方法和电子装置与流程
本发明涉及融合基因检测领域,具体而言,涉及一种判断融合基因真实性的方法和电子装置。
背景技术:
1、融合基因(fusion gene)是指由两个或更多基因的片段融合而成的新基因。在肿瘤组织中,融合基因是一种常见的基因结构异常,两个基因的片段融合在一起产生了一个新的融合基因,具有不同于原始基因的序列结构和功能。肿瘤组织中的融合基因在癌症的发生和发展过程中起着重要的作用。融合基因可以通过多种机制改变细胞的生长、分化和凋亡等关键过程,从而促进肿瘤的形成。
2、在现有技术中,可以通过传统的pcr方法对融合断点设计探针进行验证,也可以结合一定的生物信息学方法,对发生融合的蛋白结构域进行分析,再结合已发表文献对筛选出可能与该肿瘤相关的融合基因。但传统pcr验证的方法需要而外的时间成本和资金成本,另外由于临床样本多为ffpe样本,样本提取核酸的难度较大,验证结果不一定准确。通过生物信息学的方法需要一定的技术积累和经验,且具有一定的主观性,不同人鉴定的结果会有差异。
3、在现有技术中也可以利用rna测序,包括但不限于rna捕获测序,检测肿瘤组织样本中发生的融合基因。但在利用rna捕获测序检测融合基因的过程中,涉及样本提取、文库构建、上机测序及生信分析等多种环节,每个环节的异常或变动会极大影响融合基因的检测,因此此种方法的准确度较低。在现有技术中虽然也公开了一种通过对融合基因的外显子进行表达定量,通过比较融合断点两端的外显子的表达差异来判断融合基因的真阳性的方法,但在实际应用中,由于每个外显子长度不一样,不同区域探针的捕获效率不一样,导致直接对外显子定量无法准确判断断点两侧外显子的表达差异,对于融合基因的判断的准确率仍较低。
技术实现思路
1、本发明的主要目的在于提供一种判断融合基因真实性的方法和电子装置,以解决现有技术中的利用rna测序数据判断融合基因的准确率低的问题。
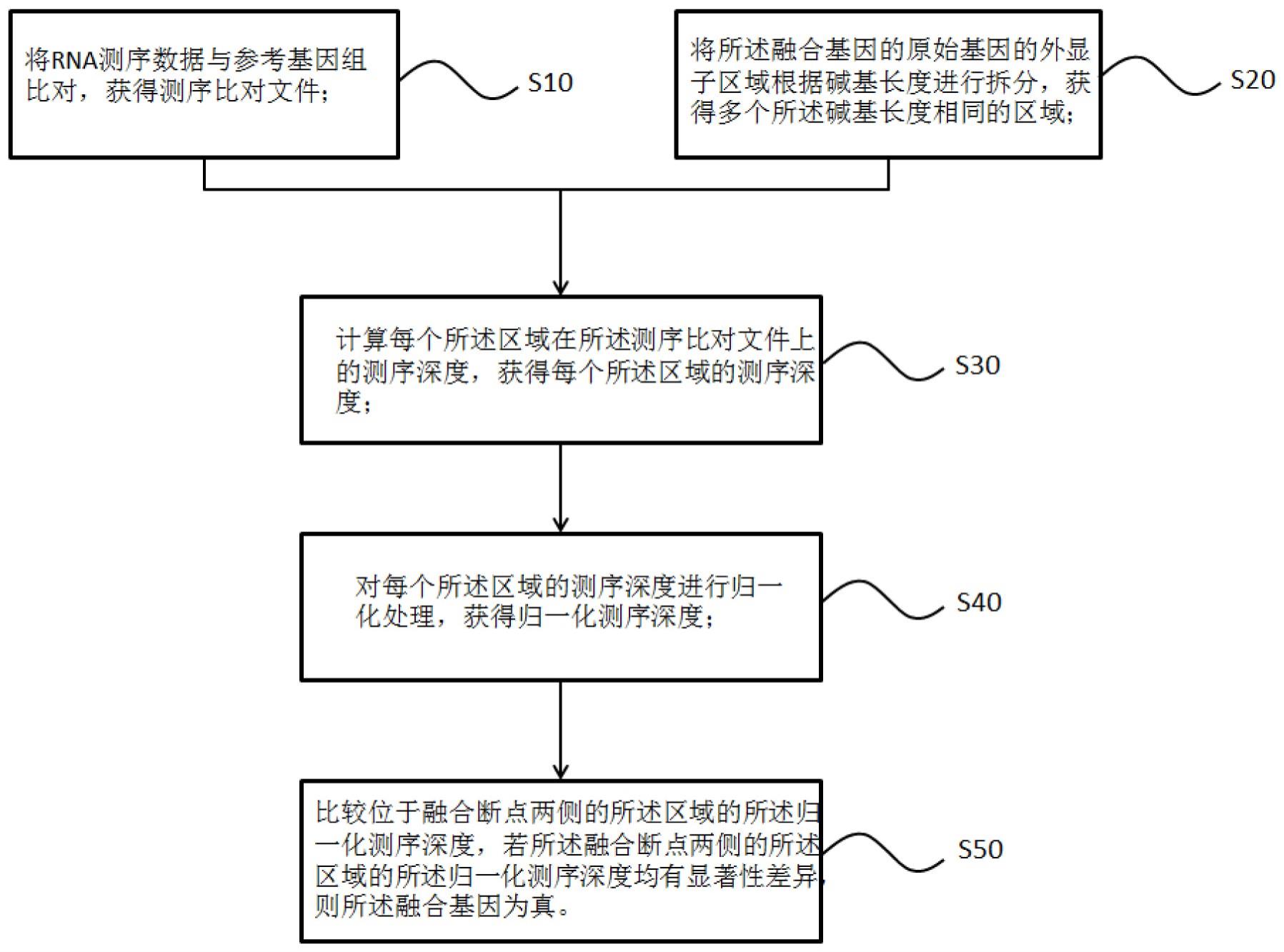
2、为了实现上述目的,根据本发明的第一个方面,提供了一种判断融合基因真实性的方法,该方法包括:a)将rna测序数据与参考基因组比对,获得测序比对文件;b)将融合基因的原始基因的外显子区域根据碱基长度进行拆分,获得多个碱基长度相同的区域;c)计算每个区域在测序比对文件上的测序深度,获得每个区域的测序深度;d)对每个区域的测序深度进行归一化处理,获得归一化测序深度;e)比较位于融合断点两侧的区域的归一化测序深度,若融合断点两侧的区域的归一化测序深度均有显著性差异,则融合基因为真。
3、进一步地,归一化处理包括:对测序深度进行测序数据量的归一化处理,获得每百万条数据量测序深度;再对每百万条数据量测序深度进行测序效率的归一化处理,获得归一化测序深度。
4、进一步地,测序数据量的归一化处理包括:获得rna测序数据的测序数据量,根据测序深度和测序数据量计算获得每百万条数据量测序深度,每百万条数据量测序深度 =测序深度 ÷ 测序数据量 × 106。
5、进一步地,测序效率的归一化处理包括:利用归一化因子对每百万条数据量测序深度进行测序效率的归一化处理,获得归一化测序深度,归一化因子包括中位数归一化因子和/或平均数归一化因子,归一化测序深度包括中位数归一化测序深度和/或平均数归一化测序深度,中位数归一化测序深度 = 每百万条数据量的测序深度 × 中位数归一化因子,平均数归一化测序深度 = 每百万条数据量的测序深度 × 平均数归一化因子。
6、进一步地,归一化因子的计算方法包括:d1)对同一样本进行多次rna捕获测序,并将测序结果分别与参考基因组比对,获得多个rna参考数据;d2)对于每个rna参考数据,计算每个区域在rna参考数据中的测序深度;d3)统计每个区域在多个rna参考数据中的测序深度,获得每个区域的测序深度的中位数和/或平均数,计算每个区域的中位数归一化因子和/或平均数归一化因子,中位数归一化因子 = 100÷ 中位数,平均数归一化因子 = 100÷ 平均数。
7、进一步地,碱基长度为8-20 bp。
8、为了实现上述目的,根据本发明的第二个方面,提供了一种判断融合基因真实性的电子装置,该电子装置包括序列比对单元、区域拆分单元、测序深度计算单元、归一化处理单元和显著性判断单元;其中,序列比对单元,用于将rna测序数据与参考基因组比对,获得测序比对文件;区域拆分单元,用于将融合基因的原始基因的外显子区域根据碱基长度进行拆分,获得多个碱基长度相同的区域;测序深度计算单元,用于计算每个区域在测序比对文件上的测序深度,获得每个区域的测序深度;归一化处理单元,用于对每个区域的测序深度进行归一化处理,获得归一化测序深度;显著性判断单元,用于比较位于融合断点两侧的区域的归一化测序深度,若融合断点两侧的区域的归一化测序深度均有显著性差异,则融合基因为真。
9、进一步地,归一化处理单元包括测序数据量归一化单元和测序效率归一化单元;其中,测序数据量归一化单元,用于对于测序深度进行测序数据量的归一化处理,获得每百万条数据量测序深度;测序效率归一化单元,用于对于每百万条数据量测序深度进行测序效率的归一化处理,获得归一化测序深度。
10、进一步地,测序数据量归一化单元包括:第一获取模块,用于获取rna测序数据的测序数据量,以及第一计算模块,用于计算每百万条数据量测序深度,每百万条数据量测序深度=测序深度 ÷ 测序数据量 × 106;测序效率归一化单元包括:第二获取模块,用于获取归一化因子,归一化因子包括中位数归一化因子和/或平均数归一化因子,以及第二计算模块,用于利用归一化因子对每百万条数据量测序深度进行测序效率的归一化处理,获得归一化测序深度,归一化测序深度包括中位数归一化测序深度和/或平均数归一化测序深度,中位数归一化测序深度 = 每百万条数据量的测序深度 × 中位数归一化因子,平均数归一化测序深度 = 每百万条数据量的测序深度 × 平均数归一化因子。
11、进一步地,测序效率归一化单元中储存有中位数归一化因子和/或平均数归一化因子,或测序效率归一化单元中包括归一化因子计算单元,归一化因子计算单元包括:获取比对模块,用于获取对同一样本进行多次rna捕获测序的测序结果,并将测序结果分别与参考基因组比对,获得多个rna参考数据;深度计算模块,用于对于每个rna参考数据,计算每个区域在rna参考数据中的测序深度;以及归一化因子统计计算模块,用于统计每个区域在多个rna参考数据中的测序深度,获得每个区域的测序深度的中位数和/或平均数,并计算每个区域的中位数归一化因子和/或平均数归一化因子,中位数归一化因子 = 100÷ 中位数,平均数归一化因子 = 100÷ 平均数。
12、进一步地,碱基长度为8-20 bp。
13、为了实现上述目的,根据本发明的第三个方面,提供了一种计算机可读储存介质,该储存介质包括存储的程序,其中,在程序运行时,控制储存介质所在设备执行上述方法。
14、为了实现上述目的,根据本发明的第四个方面,提供了一种处理器,该处理器用于运行程序,其中,程序运行上述方法。
15、应用本发明的技术方案,利用上述判断融合基因真实性的方法,通过将外显子区域拆分为特定碱基长度的区域(bin),统计每个bin的测序深度,并对测序深度进行归一化处理获得归一化测序深度,通过对融合断点两侧的bin的归一化测序深度进行显著性分析,从而实现利用生物信息学的方法准确判断融合基因的真实性,避免外显子长度、gc含量等因素影响对于融合基因判断准确性的影响。
- 还没有人留言评论。精彩留言会获得点赞!